Genetikk – er betydelige effekter dokumentert ved schizofreni?
Roar Fosse
-
Roar Fosse
Klinikk for psykisk helse og rus, Vestre Viken
roar.fosse@vestreviken.no
Genetic research on schizophrenia started out with a focus on candidate genes, but with limited success. Subsequent studies have searched for schizophrenia-associated single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) anywhere on the genome. In genome wide association studies (GWAS), SNPs with a small but statistically significant difference in prevalence between schizophrenia and normal healthy controls have been identified at 108 independent positions on DNA, accounting for 3,4 % of liability to the disorder. CNVs appear to account for an additional 1 % of liability but are unspecific for schizophrenia. Sum scores of genetic risk based on SNPs from GWAS are used to investigate associations with underlying symptom dimensions and neurobiological and cognitive traits, but substantial associations remain to be identified. The current molecular genetic evidence does not support a view of schizophrenia as strongly genetically based.
Keywords: schizophrenia, endophenotypes, single nucleotide polymorphisms, copy number variations, polygenic risk scores
Flere sentrale strukturelle endringer i hjernen ved schizofreni sammenfaller med de som ses etter alvorlig relasjonsstress i oppveksten
Hypotesen om genetisk sårbarhet eller predisposisjon for vedvarende, ikke-affektive psykoser (schizofreni) hviler på observasjoner av at risikoen for lidelsene øker proporsjonalt med hvor nær i slekt vi er med en person med en slik diagnose (Gottesman, 1991). Basert på tvillingstudier har atferdsgenetikere estimert at 44 til 87 % av variasjonen i schizofreni i befolkningen kan forklares med genetiske forskjeller (arvelighet) (Sullivan, Kendler, & Neale, 2003). En kritikk mot tvillingstudier og andre atferdsgenetiske metoder er imidlertid at de er uegnet til å skille mellom (genetisk) arv og miljø, slik at arvelighetsestimatene er ugyldige (Fosse, 2013; Fosse, Joseph, & Richardson, 2015; Joseph, 2006, 2015; Lewontin, Rose, & Kamin, 1984). Direkte studier av variasjoner i DNA-strukturen vil gi sikrere kunnskap om genetiske bidrag til schizofreni.
Frem til ca. 2008 var hovedstrategien i den molekylærgenetiske forskningen å undersøke betydningen av gener for nevrobiologiske prosesser som man antok var endret i schizofreni (kandidatgener), for eksempel genene betegnet som COMT og DRD2 for dopaminfunksjon. I innledende publikasjoner fra denne forskningen rapporterte forskerne om positive sammenhenger, men disse lot seg ikke replisere i oppfølgingsstudier. I den største studien på området frem til da undersøkte Sanders og medarbeidere (2008) variasjoner (alleler) i 14 sentrale kandidatgener for schizofreni. De fant ingen sammenheng og konkluderte: «… det finnes trolig ikke ekte sammenhenger på befolkningsnivå for de allelene som har utgjort grunnlaget for den store kandidatgenlitteraturen for disse 14 postulerte kandidatgenene for schizofreni» (s. 505). Sju år senere publiserte forskere som selv hadde vært sentrale i kandidatgenforskningen, en metaanalyse av 25 sentrale kandidatgener og kom til samme konklusjon: «Oppsummert, den empiriske evidensen støtter nå sterkt ideen om at den historiske kandidatgenlitteraturen ikke gav noen robuste og replikerbare innsikter i etiologien ved schizofreni» (Farrell et al., 2015, s. 560).
Den manglende støtten til kandidatgenene førte til en vridning i hvordan genetikk ble antatt å bidra til schizofreni (og andre psykiske lidelser). I tråd med en hypotese antydet av Ronald Fisher i 1918 og mer eksplisitt formulert av Gottesman og Shields i 1967, antok forskerne i stedet at predisposisjonen hviler på et stort antall genvarianter som hver har kun en svært liten effekt. For å identifisere slike små effekter trengte man store studier med svært mange deltakere, gjerne flere titusen. Siden man ikke lenger hadde noen klar hypotese om hvor på genomet eventuelle signifikante variasjoner fantes, inntok feltet en hypotesefri fremgangsmåte der alt tilgjengelig DNA på kromosomene ble studert. For å få til slike enorme studier ble det dannet internasjonale konsortier, der gendatabasene fra en rekke land ble slått sammen.
Problemstillingen i denne artikkelen er hvorvidt de siste årenes molekylærgenetiske forskning har dokumentert en betydelig rolle for strukturelle variasjoner på DNA for personer med schizofrenidiagnose. «Betydelig» forstås her pragmatisk som forklart varians basert på Cohens rangering av størrelsen på korrelasjonskoeffisienter (Cohen, 1992), der mindre enn 1 % forklart varians forstås som triviell, 1 til 9 % som lite, 9 til 25 % som medium og over 25 % som betydelig/stor. I tillegg vurderes om foreliggende evidens og kunnskap sannsynliggjør at videre forskning vil finne betydelige sammenhenger.
For å belyse problemstillingen gjennomgås funn fra den molekylærgenetiske forskningen på schizofreni, med utgangspunkt i enkeltnukleotidpolymorfismer eller enkeltbasevariasjoner (single nucleotide polymorphism, SNP). En SNP innebærer en variasjon mellom folk i baseparene på en posisjon på DNA, som ved at noen har adenin-thymin, mens andre har guanin-cytosin på en gitt posisjon på et kromosom (figur 1). Ca. 1 % av de over tre milliarder baseparene på DNA varierer på denne måten, mens resten er like for alle (fikserte). SNP-er kan forekomme i eller utenfor de 20 000–25 000 genene på DNA, der et gen består av fra flere tusen til flere millioner basepar.
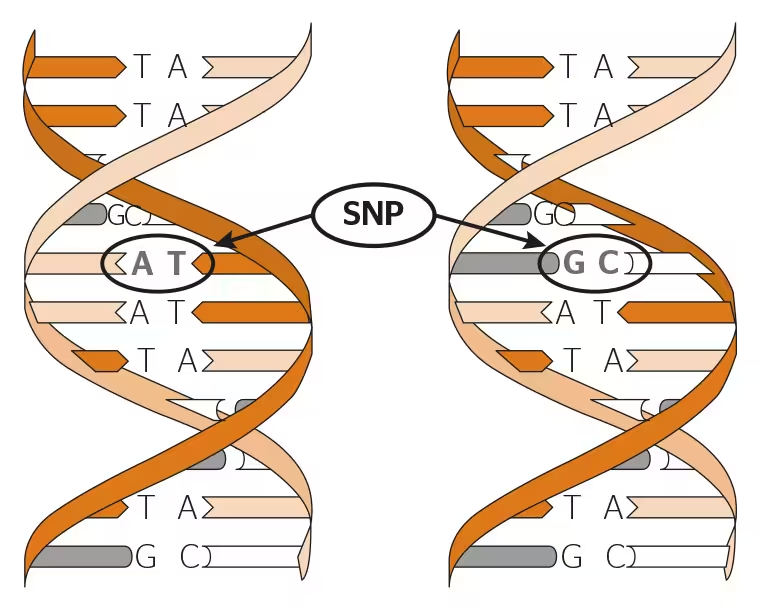
Figur 1. Enkeltnukleotidpolymorfisme (single nucleotide polymorphism, SNP)
En SNP ses når et basepar på en gitt posisjon på et kromosom kan variere mellom folk, dvs. ved at noen har adenin-thymin (A-T), mens andre har guanin-cytosin (G-C) på denne posisjonen. Figuren er lastet ned fra http://www.viagenefertility.com/Available-PGD-Technologies.php.
Dernest gjennomgås funn fra studier av kopitallsvariasjoner (copy number variation, CNV). CNV sikter til variasjon i forekomsten av en gitt sekvens på DNA hos et individ, bestående av fra tusen basepar og oppover (Barøy, Misceo, & Frengen, 2008; Stankiewicz & Lupski, 2010). CNV-er forekommer som duplisering (flere kopier av en sekvens), innsetning (et segment er kopiert og satt inn et annet sted på DNA) og delesjon (en sekvens er fjernet). Se ellers figur 2.
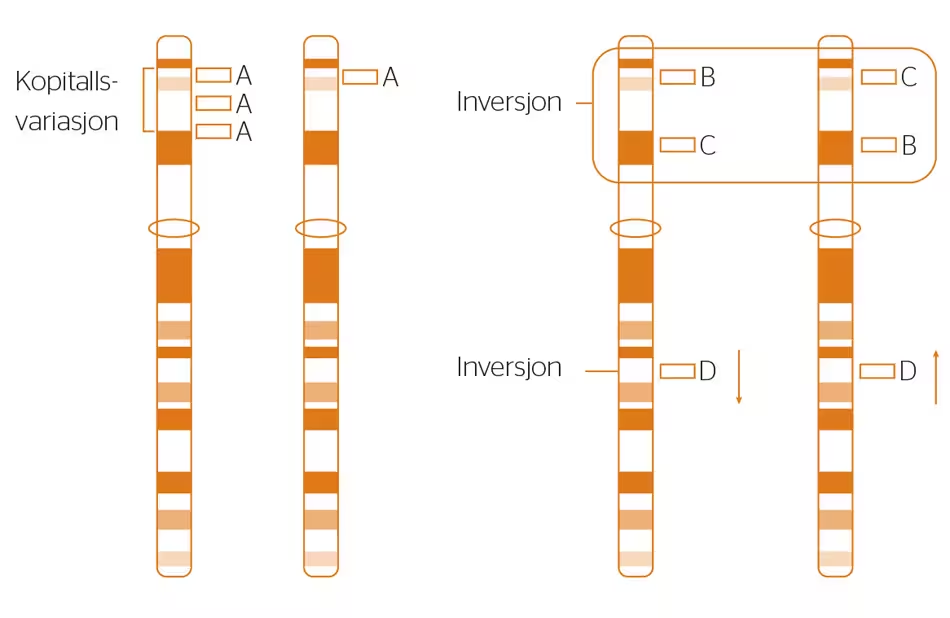
Figur 2. Kopitallsvariasjoner (copy number variations, CNV)
Duplisering av det genomiske området A (venstre) og inversjon av området B-C (høyre). Hentet fra Barøy, Misceo og Frengen (2008).
Etter dette gjennomgås forskning på sammenhengen mellom SNP-er og såkalte endofenotyper – enkeltsymptomer, nevrobiologisk struktur/funksjon og kognitive funksjoner som er endret ved schizofreni. Et utgangspunkt for denne forskningen er den uttalte variasjonen i symptombilde som ses blant personer diagnostisert med schizofreni, som erkjent av Bleuler allerede i 1911 (Bleuler & Bleuler, 1986). Endofenotyper kan antas å ligge mellom årsaksfaktorene og schizofreni og være tydeligere knyttet til genetikk enn det den nokså heterogene kategorien schizofreni er (Insel & Cuthbert, 2009).
Metode
Gjennomgangen av de siste årenes molekylærgenetiske forskning fokuserer på de største studiene som er gjennomført i regi av internasjonale konsortier og publisert siden 2009. Dette valget er begrunnet i at funn fra mindre studier er ureliable, og at forskningsfeltet selv har argumentert for at det er nødvendig med store, akkumulerte databaser fra samarbeidende forskergrupper for å drive feltet videre. Studiene ble identifisert ved hjelp av oversiktsartikler og søk på PubMed med ulike kombinasjoner av søkeordene genome-wide association, GWA*, psychosis, schizophrenia, genetic*, DNA, single nucleotide polymorphism, SNP, copy number variation og CNV.
Studier av sammenhengen mellom SNP-er og fenotyper ble identifisert ved søk i PubMed med kombinasjoner av følgende søkeord: psychosis, schizophrenia, single nucleotide polymorphism, SNP, polygenic, risk score, PGRS, PRS, phenotype, endophenotype, positive symptoms, negative symptoms, hallucinations, disorganization, delusions, cognitive, neurocognitive, prefrontal, frontal, hippocampus, amygdala, regional, activation. I tillegg ble referanselistene i identifiserte artikler gjennomgått. Inkludert er artikler publisert frem til august 2016.
Resultater
Enkeltnukleotidpolymorfismer
Det internasjonale schizofrenikonsortiet publiserte i 2009 den første megastore genomvide assosiasjonsstudien («genome wide association study», GWAS) for schizofreni, med 3322 pasienter og 3587 normalt friske kontrollpersoner (International Schizophrenia Consortium, 2009). Konsortiet undersøkte forskjeller i ca. 1 million SNP-er mellom de to deltakergruppene. De fant kun ett område på DNA med en SNP som forekom signifikant forskjellig på genomnivå, definert ved en p-verdi mindre enn 5 x 10–8. Et slikt strengt signifikansnivå er nødvendig for å beskytte mot falske positive funn (type I statistisk feil), noe som ellers er overhengende på grunn av det store antallet tester i en GWAS (ekvivalent med antall SNP-er som undersøkes). SNP-en som var signifikant i konsortiets studie, lå i det såkalte major histocompatibility complex (MHC), et stort område på kromosom 6 som strekker seg over 3,6 millioner basepar og er knyttet til immunsystemet. Studien indikerte samtidig at det neppe finnes noen SNP-er som er nær unike for schizofreni; sannsynligvis finnes ingen schizofreni-assosiert SNP med odds ratio større enn 1,3, det vil si som forekommer 30 % oftere (eller sjeldnere) i en gruppe med schizofrenidiagnose enn hos normalt friske personer (Wray et al., 2014).
I et neste analyseskritt i konsortiets 2009-studie antok forskerne at selv om kun én SNP nådde det nødvendige genom-overgripende signifikansnivået for schizofreni, så kunne det likevel foreligge mange, kanskje flere tusen, med en ørliten sammenheng som ville vært signifikante hvis utvalget hadde vært større. For å undersøke dette tok de utgangspunkt i de mannlige pasientene i utvalget og lagde en sumskåre basert på 37 655 SNP-er som nådde et svært liberalt signifikansnivå på p = 0,5 (hvilket tilsier at en stor andel var falske positiver). Slike sumskårer har siden blitt betegnet som polygenetiske risikoskårer (PGRS), der skåren beregnes ved at hver SNP vektes med dens effektstørrelse (den naturlige logaritmen av odds ratioen til SNP-en). Hver person i et nytt testutvalg skåres da på denne summerte skalaen. Schizofrenikonsortiet (2009) fant at sumskåren som de etablerte basert på menn, var signifikant (p = 9,4 x 10–19) knyttet til schizofreni blant kvinner, som da utgjorde testgruppen i studien, og at den her kunne forklare ca. 3 % av variansen i schizofreni (Nagelkerkes R 2). De konkluderte med at funnet støttet antakelsen om schizofreni som en polygenetisk lidelse, og at kommende studier med enda større deltakergrupper ville avdekke flere genomvid signifikante (p < 5 x 10–8) SNP-er.
Schizofrenikonsortiets (2009) hypotese ble støttet av funn fra en enda større GWAS fra et annet konsortium (SGENE) som ble publisert samme år, med 12 945 deltakere med schizofrenidiagnose og 34 591 normalt friske personer (Stefansson et al., 2009). Genomvid signifikans ble her nådd for sju SNP-er, hver med odds ratio på mellom 1,15 og 1,24. Fem av dem lå i MHC-området for immunsystemet. To år senere publiserte «Schizophrenia Working group of the Psychiatric Genomics Consortium» (2011) en GWAS med 17 836 pasienter og 33 859 normalt friske kontrollpersoner. Også nå identifiserte man sammenhenger med schizofreni i MHC-området, i tillegg til i seks andre områder. Disse nye SNP-ene hadde en enda mindre odds ratio: ~ 1,1.
Disse likhetene stiller spørsmålstegn ved om genetikk behøves for å forklare sentrale nevrobiologiske endringer ved schizofreni
I 2014 ble den til nå siste og største studien publisert, der deltakerantallet var økt til 37 000 pasienter med schizofrenidiagnose og 113 000 normalt friske mennesker (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Her ble det rapportert om 128 signifikante SNP-er på 108 ulike områder på DNA. Blant de 108 områdene var igjen immunsystemet (MHC) implisert, i tillegg til områder knyttet til glutamatfunksjon og synaptisk plastisitet, kalsiumsignalisering og dopaminfunksjon. De signifikante SNP-ene kunne til sammen forklare 3,4 % av tilbøyeligheten til schizofreni, med gjennomsnittlig forklart varians per SNP på 0,03 %. Sju prosent av variasjonen i schizofreni i utvalget kunne forklares med en PGRS basert på flere tusen ikke-signifikant assosierte SNP-er.
Et viktig aspekt ved disse funnene er at genvarianter som er genomvidt signifikante, er tilnærmet like vanlige blant normalt friske mennesker som blant de med schizofrenidiagnose. I 2014-studien fra schizofrenikonsortiet forekom topp-SNP-en hos 86,4 % av deltakerne med schizofreni og 85,0 % av de normalt friske. Den andrerangerte forekom hos henholdsvis 17,5 % og 19,1 % (tabell 1). To ledende genforskere kommenterte dette slik: «Vi bærer alle på risikovarianter for schizofreni, og de aller fleste av oss bærer på nokså mange av dem. Når det gjelder genetisk risiko er det ingen ‘dem’ og ‘oss’, kun subtile skygger av grått» (Kendler & O’Donovan, 2014 s. 1320).
Tabell 1. De 10 mest signifikante SNP-ene i 2014-studien til «Schizophrenia Working Group of the Psychiatric Genomics Consortium»
Rangering |
Kromosom |
SNP |
Allele* |
Schizofreni ( %) |
Normal-gruppe (%) |
Oddsratio1 |
|---|---|---|---|---|---|---|
1 |
6 |
rs115329265 |
AG |
86,4 |
85,0 |
1,21 |
2 |
1 |
rs1702294 |
TC |
17,5 |
19,1 |
0,89 |
3 |
10 |
rs11191419 |
AT |
33,7 |
36,0 |
0,91 |
4 |
12 |
rs2007044 |
AG |
60,2 |
62,4 |
0,91 |
5 |
8 |
rs4129585 |
AC |
44,7 |
42,4 |
1,08 |
6 |
4 |
rs35518360 |
AT |
90,9 |
92,2 |
0,87 |
7 |
7 |
chr7_2025096 |
Innsetning |
40,5 |
42,3 |
0,92 |
8 |
5 |
rs4391122 |
AG |
50,5 |
53,2 |
0,92 |
9 |
12 |
rs2851447 |
CG |
72,3 |
74,1 |
0,91 |
10 |
2 |
chr2_20082537 |
Innsetning |
74,1 |
75,4 |
0,91 |
1 Odds-ratio i discovery-utvalget, hentet fra Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014), supplementær tabell 2, s. 22–25, se http://www.nature.com/nature/journal/v511/n7510/extref/nature13595-s1.pdf. *A = Adenin, G = Guanin, T = Thymin, C = Cytosin. | ||||||
Kopitallsvariasjoner (CNV)
En liten, men signifikant økning i CNV-er er identifisert ved schizofreni (Malhotra & Sebat, 2012; Rees et al., 2014). I en studie med 6882 pasienter og 6316 normalt friske personer rapporterte Rees og medarbeidere (2014) at de 15 CNV-ene som var sterkest assosiert med schizofreni i forutgående studier, til sammen forekom 171 ganger i schizofrenigruppen (hos 2,5 % av pasientene) sammenliknet med 58 ganger i kontrollgruppen (0,9 %). I den hittil største studien, en metaanalyse med 21 094 pasienter og 20 227 normalt friske personer, fant forskerne CNV-er på åtte områder på DNA som forekom signifikant forskjellig mellom gruppene (Marshall et al., 2016). Disse åtte CNV-ene kunne forklare 0,85 % av tilbøyeligheten til schizofreni i utvalget. Hver av de impliserte CNV-ene forekom svært sjelden selv ved schizofreni.
Endofenotyper
Fordi schizofrenidiagnosen er heterogen, kan det være at genetiske bidrag vil fremstå som tydeligere for underliggende endofenotyper – enkeltsymptomer og nevropsykologiske avvik. En rekke studier har undersøkt dette ved å ta utgangspunkt i enkeltstående, antatt viktige genvarianter. Ingen konsistente funn er imidlertid rapportert (Gurung & Prata, 2015). En utfordring for studier som tar utgangspunkt i enkeltstående genvarianter, er indikasjonen fra GWAS om at hver enkelt variant kun har en svært liten effekt. Farrel og medarbeidere (2015) estimerte at selv med 1000 pasienter og 1000 kontrollpersoner har slike studier en styrke på kun 0,03 %. Dette betyr at det vil være vanskelig å påvise sammenhenger for enkeltvarianter, med en overveldende fare for falske positive funn. Det har ikke forenklet denne utfordringen at slike studier ofte har tatt utgangspunkt i kandidatgener, en fremgangsmåte som hadde liten suksess (Greenwood et al., 2016; Yeo et al., 2014).
Fra rundt 2012 dreide derfor forskningen på endofenotyper mot å bruke PGRS slik disse er utledet fra GWAS. Dette gir mening også fordi sentrale endofenotyper som kognitive funksjoner har et uttalt polygenetisk grunnlag med små effekter for hver variant (Mark & Toulopoulou, 2016). Nedenfor oppsummeres funnene fra denne forskningen, og de er mer detaljert gjengitt i tabell 2, 3 og 4. I disse studiene har schizofreni-PGRS oftest vært basert på et stort antall (hundrevis eller flere tusen) SNP-er som har vært nominelt (heller enn statistisk signifikant) knyttet til schizofreni i GWAS. Dette gjør PGRS til en usikker størrelse og dermed fremgangsmåten noe tentativ.
Tabell 2. Sammenheng mellom SNP-baserte polygenetiske risikoskårer og psykosesymptomer
Studie |
Deltakere |
Grunnlag for PGRS |
Utfallsmål |
Sammenheng med PGRS |
|---|---|---|---|---|
Derks et al. 2012 |
314 SZ og 148 normalt friske |
PGC-2011 |
Positive og negative psykose-symptomer, disorganisering, mani |
Signifikante assosiasjoner, men skyldtes generelt høyere PGRS i SZ-gruppen. Ingen sammenheng innenfor hver gruppe |
Fanous et al. 2012 |
2454 SZ |
6715 fra PGC |
Positive og negative psykose-symptomer, disorganisering (tankeforstyrrelser, bisarr atferd), humør |
Signifikant assosiert med (kun) disorganiserte symptomer, R2 = 2,2 % |
Sieradzka et al. 2014 |
2152 normalt friske 16-åringer |
PGC-2011 |
Paranoia, hallusinasjoner, kognitiv disorganisering, grandiositet, anhedoni |
Ingen sammenhenger |
Zammit et al. 2014 |
3483 normalt friske ungdommer |
(i) PGC-2011 og (ii) 17 signifikante SNP-er fra PGC 2011 |
Typiske psykoseopplevelser som hallusinasjoner, vrangforestillinger og tankeforstyrrelser |
Ingen sammenheng basert på (i). For (ii) lavere psykosesymptomer for de med høyest PGRS |
Jones et al. 2016 |
9912 normalt friske ungdommer |
PGC-2014 |
Positive symptomer (hallusinasjoner, vrangforestillinger og tankeforstyrrelser) og negative symptomer (apati, anergi, asosialitet) |
Trend for positive symptomer, signifikant sammenheng med negative symptomer – R2 = 0,7 % |
SZ = Schizofreni. PGRS = Polygenetisk risikoskåre for schizofreni utledet fra GWAS. PGC = Psychiatric genetic consortium. SNP = Single nucleotide polymorphism / enkeltnukleotidpolymorfisme. R2 = Nagelkerkes pseudo R2 (forklart varians) | ||||
Tabell 3. Sammenheng mellom SNP-baserte polygenetiske risikoskårer og hjernestruktur
Studie |
Deltakere |
Grunnlag for PGRS |
Utfallsmål |
Sammenheng med PGRS |
|---|---|---|---|---|
Terwisscha van Scheltinga et al. 2013 |
152 SZ og 142 normalt friske |
Deler av PGC-2011 |
Totalt volum på (i) hele hjernen, (ii) grå masse, (iii) hvit masse |
Signifikant sammenheng med (mindre) totalt hjernevolum (R2 = 4,8 %) og totalt hvit masse (R2 = 5,1 %) |
Papiol et al. 2014 |
122 normalt friske |
PGC-2011 |
Totalt volum på (i) hele hjernen, (ii) grå masse, (iii) hvit masse |
Ingen sammenhenger |
Van der Auwera et al. 2015 |
1470 normalt friske |
PGC-2014 |
Totalt volum på (i) hele hjernen, (ii) grå masse, (iii) hvit masse og volum lokalt hvor som helst |
Ingen sammenhenger |
Voineskos et al. 2016 |
565 normalt friske |
91 SNP-er fra PGC-2014 |
Tykkelse på frontal og temporal hjernebark, frontotemporal og interhemisfærisk diffusjon |
Ingen sammenhenger |
Oertel-Knøchel 2015 |
94 SZ og normalt friske |
7 SNP-er fra PGC 2011 |
Hvit materie |
Sammenheng med hvit materie i hele gruppen, R2 = 4,4 %. Ingen korrelasjon innenfor SZ-gruppen |
Liu et al. 2016 |
315 normalt friske |
PGC-2014 |
Kortikal gyrifisering (grad av kortikal folding) |
Sammenheng med gyrifisering i parietallappene, R2 =1–2 % |
Franke et al. 2016 |
11 840 fra befolkningen |
PGC-2014 |
Subortikale områder inkl. hippocampus, amygdala, thalamus, nucleus accumbens |
Ingen sammenhenger |
SZ = Schizofreni. PGRS = Polygenetisk risikoskåre for schizofreni utledet fra GWAS. PGC = Psychiatric genetic consortium. SNP = Single nucleotide polymorphism / enkeltnukleotidpolymorfisme. R2 = Nagelkerkes pseudo R2 | ||||
Tabell 4. Sammenheng mellom SNP-baserte polygenetiske risikoskårer, kognitiv funksjon og hjerneaktivering
Studie |
Deltakere |
Grunnlag for PGRS |
Utfallsmål |
Sammenheng med PGRS |
|---|---|---|---|---|
Hjerneaktivering ved kognitiv testing | ||||
Walton et al. 2013 |
77 SZ og 99 normalt friske |
41 SNP-er fra PGC per februar 2010 |
Prefrontal aktivering v/ arbeidshukommelsestest |
Sammenheng med mer aktivering i venstre DLPFC. R2 maks = 3,6 % |
Walton et al. 2014 |
255 SZ |
600 SNP-er fra 3322 pasienter |
Prefrontal aktivering v/ arbeidshukommelsestest |
Signifikant assosiasjon (mindre aktivering) i venstre DLPFC og ventrolaterale PFC. Maks R2 = 4,3 % |
Kauppi et al. 2015 |
63 SZ og 118 normalt friske |
PGC-2014 |
Prefrontal aktivering v/ arbeidshukommelsestest |
Assosiert med mindre aktivering i øvre PFC og inferior frontal gyrus. R2 = 2,8–3,5 % |
Kognitiv funksjon | ||||
McIntosh 2013 |
1454 normalt friske |
PGC-2011 |
IQ/ kognitiv evne ved alder 70 og endring i IQ fra 11 år til 70 år |
Assosiert med begge utfallsmålene, R2 = 0,3–0,9 % |
Terwisscha van Scheltinga et al. 2013 |
350 SZ og 322 normalt friske |
Deler av PGC-2011 |
Intelligens (WAIS) |
Assosiert med gruppe, men ikke med intelligens v/ kontroll for gruppe |
Yeo et al. 2014 |
50 SZ og 86 normalt friske |
41 SNP-er fra PGC per februar 2010 |
Eksekutivfunksjon: planlegging, flyt og inhibisjon |
Ingen sammenheng |
Lencz et al. 2014 |
~5000 normalt friske |
PGC-2011 |
Generell kognitiv evne |
Signifikant sammenheng, R2 < 2 % |
Hatzimanolis et al. 2015 |
1079 normalt friske menn |
PGC-2014 |
11 kognitive funksjoner inklusive arbeidshukommelse, vedvarende oppmerksomhet og ikke-verbal IQ (WAIS) |
Ingen sammenheng som nådde adekvat signifikansnivå |
Liebers et al. 2016 |
8618 normalt friske eldre |
PGC-2014 |
Korte/enkle tester av verbal korttidsminne, oppmerksomhet og språk |
Signifikant sammenheng med oppmerksomhet (R2 = 0,08 %) og verbalt korttidsminne (R2 = 0,02 %) |
Hubbard et al. 2016 |
> 5000 normalt friske barn, 8 år |
PGC-2014 |
IQ, oppmerksomhet, prosesserings-hastighet, arbeidshukommelse, problemløsning, sosial kognisjon |
Signifikant sammenheng med IQ (R2 = 0,19 %), men ikke med andre kognitive mål |
Lancaster et al. 2016 |
100 normalt friske |
PGC-2014 |
Fem betingelser for belønningsbasert probabilistisk beslutningstaking |
Sammenheng for én av fem testbetingelser med lavere aktivering i høyre frontallapp (p=0,048) og venstre ventrale striatum (p=0,036). R2 ikke oppgitt |
SZ = Schizofreni. PGRS = Polygenetisk risikoskåre for schizofreni utledet fra GWAS. PGC = Psychiatric genetic consortium. SNP = Single nucleotide polymorphism/ enkeltnukleotidpolymorfisme. R2 = Nagelkerkes pseudo R2 | ||||
Studier som har undersøkt schizofreni-PGRS opp mot positive psykosesymptomer (hallusinasjoner og vrangforestillinger), har ikke identifisert de forventede sammenhengene hverken i pasientgrupper eller generelle befolkningsgrupper (tabell 2). Den største av disse studiene (Jones et al., 2016) fant snarere evidens for at ungdommer med høy PGRS hadde mindre psykosesymptomer enn andre ungdommer. Derimot er PGRS funnet å være noe knyttet til negative symptomer som apati, asosialitet og disorganisering, selv om sammenhengen er svært svak, med en forklart varians i disse studiene på mellom 0,7 % og 2,2 %.
En sentral kategori endofenotyper ved schizofreni er hjernestruktur. Særlig ses redusert størrelse på hippocampus, amygdala, thalamus, nucleus accumbens (dopaminsystemet) og store deler av prefrontal korteks i lidelsen (Fusar-Poli & Meyer-Lindenberg, 2016). Den største studien om PGRS og hjernestruktur ved schizofreni ble publisert i 2016, med 11 840 deltakere fra til sammen 35 land (Franke et al., 2016). Studien fant ingen sammenheng mellom PGRS og størrelsen på noe subkortikalt område, inklusive hippocampus, amygdala, nucleus accumbens og thalamus. Forfatterne konkluderte med at funnene ikke støttet hypotesen om at endringer i subkortikale hjerneområder ved schizofreni har å gjøre med genvariasjoner. Ingen konsistent sammenheng er heller funnet for PGRS med grove mål på totalt hjernevolum og hvit materie, og det er ikke observert noen sammenheng med total grå substans eller med tykkelsen på hjernebarken i frontal- og temporallappene (Oertel-Knochel et al., 2015; Terwisscha van Scheltinga, Bakker, van Haren, Derks, Buizer-Voskamp, Boos, et al., 2013; Voineskos et al., 2016)(tabell 3).
En liten håndfull studier har undersøkt om PGRS er knyttet til aktiveringsnivå i hjernen ved prestasjon på tester for arbeidshukommelse. Funnene er sprikende; PGRS er rapportert å være knyttet til både høyere og lavere aktiveringsnivå. Det er heller ikke rapportert noen sterk eller konsistent sammenheng mellom PGRS og ulike kognitive funksjoner (tabell 4).
Diskusjon
Mens genkandidatforskningen ikke gav noen konsistente funn, har den påfølgende og mer hypotesefrie forskningen indikert at en liten andel (3–7 %) av variasjonen i schizofreni i befolkningen kan bero på vanlig forekommende SNP-er på over hundre områder på DNA, der hver variant har en beskjeden sammenheng. I tillegg synes sjeldne CNV-er å være assosiert med rundt 1 % av variasjonen i schizofreni. Forskning på mer avgrensede endofenotyper har foreløpig gitt inkonsistente funn, med ingen eller kun små sammenhenger mellom PGRS og spesifikke psykosesymptomer (R2 fra 0–2 %), nevrobiologisk struktur eller funksjon (R2 fra 0–2 %) og kognitiv funksjon (R2 fra 0–4 %).
Stress-basert mutagenese kan være en viktig adaptiv prosess og en evolusjonsbasert respons for raskt å tilpasse seg endringer i miljøet
Genforskere antar at ved ytterligere å øke antall deltakere i forskningen kan det identifiseres flere DNA-varianter med en assosiasjon til schizofreni, særlig SNP-er (Andreassen et al., 2013). Slike nye varianter vil ha en enda mer beskjeden sammenheng med lidelsen enn de som er indikert frem til nå. Samtidig, mens genforskningen i hovedsak har søkt etter uavhengige og additive effekter av ulike DNA-varianter, antas det at interaksjonen mellom ulike varianter også kan spille en rolle (epistase) (Wei, Hemani, & Haley, 2014). Den nyeste store GWAS fra psykiatrikonsortiet fant imidlertid ingen indikasjon på slike interaksjonseffekter (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Det er videre mulig at bidrag fra DNA-variasjon vil bli tydeliggjort når dette studeres innenfor et genetikk-miljø interaksjonsperspektiv (Daskalakis & Binder, 2015).
Genforskere antar som regel at en akkumulering av schizofreni-assosierte SNP-er kan fremme patologi, som illustrert ved at disse gjerne betegnes som «sykdomsgener». Foreløpig er det ikke empirisk støtte til en slik forståelse, der en alternativ mulighet er at akkumulering av schizofreni-assosierte strukturvarianter snarere bidrar til variasjon i ulike trekk som faller innenfor normalområdet. Dette kan være forenlig med rapporterte sammenhenger mellom schizofreni-PGRS og det å ha et kreativt yrke, personlighetstrekk og variasjon i høyde (Bacanu, Chen, & Kendler, 2013; Gale et al., 2016; Power et al., 2015), selv om også disse sammenhengene er marginale.
CNV-ene som er statistisk assosiert med schizofreni, forekommer oftere også ved andre lidelser, som ADHD, bipolar lidelse, autisme, epilepsi, hjertelidelser, autoimmune lidelser og kreft (Malhotra & Sebat, 2012; Rees et al., 2014). I tillegg er de assosiert med abnormale ansiktstrekk, overvekt, lav høyde og vansker med motorisk koordinasjon og språklig artikulering (Bassett et al., 1998; Inoue, Natsuyama, & Miyaoka, 2014; Mulle et al., 2013) og med svekket IQ og kognitiv funksjon (Stefansson et al., 2014). CNV-en som er sterkest assosiert med schizofreni, kalt «22q11.2 delesjonssyndrom», er også identifisert ved over 180 andre kliniske vansker som omfatter så godt som alle organer og systemer i kroppen (Jonas, Montojo, & Bearden, 2014; Shprintzen, 2008). Disse andre kliniske vanskene knyttet til 22q11.2, og også de som er knyttet til andre schizofreni-assosierte CNV-er, fører ofte til psykososiale vansker, som vedvarende sosial eksklusjon, nederlagsfølelser og institusjonsbehandling (se Plaks et al., 2010). Generelle belastninger, psykososiale vansker og stress kan gjøre at disse CNV-ene er assosiert med schizofreni.
Annen forskning kan tas til inntekt for begrensninger i hvilke sammenhenger som kan avdekkes mellom variasjoner i DNA-strukturen og schizofreni. Sentralt i dette er at DNA gjør lite (transkriberer proteiner) alene, sett isolert fra de cellulære omgivelsene. Gentranskripsjonen reguleres av en rekke prosesser i cellen, som formen på (foldingen av) proteiner, mobile reseptorer (f.eks. glukokortikoidreseptoren) og epigenetiske prosesser som cytosinbasemetylering og histonmodifiseringer. De genregulerende prosessene er høyst sensitive for ekstracellulær påvirkning, herunder stress i miljøet (Kundakovic & Champagne, 2015; McEwen et al., 2015; Weaver, 2009). I tråd med dette er det veldokumentert at schizofreni er knyttet til en forhøyet forekomst av stress, særlig relasjonsstress. En metaanalyse indikerte at mellom 16 og 47 % (snitt 33 %) av schizofreni i befolkningen kan forklares med vanlige former for psykososialt stress i oppveksten; seksuelt, fysisk og emosjonelt overgrep, neglisjering, mobbing og død hos foreldrene (Varese et al., 2012). Befolkningsstudier har indikert sterke dose-respons-sammenhenger, der de høyeste stress-dosene er knyttet til opp mot 190 ganger økt forekomst av psykosesymptomer (Shevlin, Dorahy, & Adamson, 2007). Enkeltstudier indikerer at også andre former for miljøerfaringer er knyttet til psykoser, som prenatalt stress, fattigdom, å vokse opp i en by, å være vitne til vold mellom foreldrene, dysfunksjonelt foreldreskap – særlig emosjonsfattig overkontroll, diskriminering og rasisme, migrasjon, krigstraumer og cannabismisbruk i oppveksten (for referanser, se Read, Bentall, & Fosse, 2009). I motsetning til genetikk tilsier evidensen som nå foreligger, at miljøerfaringer spiller en betydelig rolle for utviklingen av schizofreni.
De begrensede sammenhengene som er identifisert mellom schizofreni-PGRS og nevrobiologiske fenotyper, står i kontrast til at flere sentrale strukturelle endringer i hjernen ved schizofreni sammenfaller med de som ses etter alvorlig relasjonsstress i oppveksten. Disse likhetene inkluderer redusert størrelse på hippocampus, amygdala, insula og den fremre hjernebarken, herunder tap av dendritter på pyramidecellene og redusert antall og aktivitet i internevroner som inneholder parvalbumin, samt redusert utslipp av dopamin i prefrontal korteks kombinert med økte dopaminnivåer i striatum (se Read, Fosse, Moskowitz, & Perry, 2014 for en oversikt). Disse likhetene setter spørsmålstegn ved om genetikk behøves for å forklare sentrale nevrobiologiske endringer ved schizofreni.
Det er i tillegg et åpent og lite undersøkt spørsmål om noen av de indikerte, små assosiasjonene mellom DNA-variasjoner og schizofreni reflekterer stressinduserte mutasjoner. For eksempel, mens CNV de novo-mutasjoner forekommer noe hyppigere ved schizofreni (Malhotra et al., 2011), er slike mutasjoner indikert å følge av cellulært stress, så som oksidativt stress som gjerne ses etter psykososialt stress (Fonville, Ward, & Mittelman, 2011; Maharjan & Ferenci, 2015; Melis et al., 2013). Stress-basert mutagenese kan være en viktig adaptiv prosess og en evolusjonsbasert respons for raskt å tilpasse seg endringer i miljøet (Galhardo, Hastings, & Rosenberg, 2007; Rosenberg, Shee, Frisch, & Hastings, 2012). En kan forvente å finne økte CNV-rater i grupper som har erfart mye stress, som personer med schizofrenidiagnose. Miljøerfaringer kan bidra også til andre typer mutasjoner, som punktmutasjon av cytosinbaser og dermed SNP-er (Pfeifer, 2006). Det gjenstår imidlertid å forske på sammenhenger mellom stresseksponering i oppveksten og mutagenese ved psykiske lidelser.
Konklusjon
Genforskningen har indikert at en rekke DNA-varianter spredt utover kromosomene er statistisk assosiert med schizofreni. Den forklarte variansen er imidlertid beskjeden, og det gjenstår å identifisere betydelige sammenhenger. Det er ingen uenighet om at de små sammenhengene som er indikert, mangler praktisk betydning for forebygging og behandling. Det er uklart om videre genforskning vil endre på dette, selv om ytterligere større deltakergrupper kan forventes å identifisere flere SNP-er og gi en noe økt forklart varians.
Referanser
Andreassen, O. A., Djurovic, S., Thompson, W. K., Schork, A. J., Kendler, K. S., O’Donovan, M. C., … Dale, A. M. (2013). Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet, 92(2), 197–209. doi: 10.1016/j.ajhg.2013.01.001
Bacanu, S. A., Chen, X., & Kendler, K. S. (2013). The genetic overlap between schizophrenia and height. Schizophr Res, 151(1–3), 226–228. doi: 10.1016/j.schres.2013.10.016
Barøy, T., Misceo, D., & Frengen, E. (2008). Strukturell variasjon i genomet bidrar til variasjon i egenskaper. Tidsskr Nor Lægeforen, 128(17), 1951–1955.
Bassett, A. S., Hodgkinson, K., Chow, E. W., Correia, S., Scutt, L. E., & Weksberg, R. (1998). 22q11 deletion syndrome in adults with schizophrenia. Am J Med Genet, 81(4), 328.
Bleuler, M., & Bleuler, R. (1986). Dementia praecox oder die Gruppe der Schizophrenien: Eugen Bleuler. Br J Psychiatry, 149, 661–662.
Cohen, J. (1992). A power primer. Psychol Bull, 112(1), 155–159.
Daskalakis, N. P., & Binder, E. B. (2015). Schizophrenia in the spectrum of gene-stress interactions: the FKBP5 example. Schizophr Bull, 41(2), 323–329. doi: 10.1093/schbul/sbu189
Derks, E. M., Vorstman, J. A., Ripke, S., Kahn, R. S., Schizophrenia Psychiatric Genomic, C., & Ophoff, R. A. (2012). Investigation of the genetic association between quantitative measures of psychosis and schizophrenia: a polygenic risk score analysis. PLoS One, 7(6), e37852. doi: 10.1371/journal.pone.0037852
Fanous, A. H., Zhou, B., Aggen, S. H., Bergen, S. E., Amdur, R. L., Duan, J., … Schizophrenia Psychiatric Genome-Wide Association Study, C. (2012). Genome-wide association study of clinical dimensions of schizophrenia: polygenic effect on disorganized symptoms. Am J Psychiatry, 169(12), 1309–1317. doi: 10.1176/appi.ajp.2012.12020218
Farrell, M. S., Werge, T., Sklar, P., Owen, M. J., Ophoff, R. A., O’Donovan, M. C., … Sullivan, P. F. (2015). Evaluating historical candidate genes for schizophrenia. Mol Psychiatry, 20(5), 555–562. doi: 10.1038/mp.2015.16
Fisher, R. (1918). The Correlation between Relatives on the Supposition of Mendelian Inheritance Philosophical Transactions of the Royal Society of Edinburgh, 52, 399–433.
Fonville, N. C., Ward, R. M., & Mittelman, D. (2011). Stress-induced modulators of repeat instability and genome evolution. J Mol Microbiol Biotechnol, 21(1–2), 36–44. doi: 10.1159/000332748
Fosse, R. (2013). Psykoseforståelse: et kritisk blikk på tvillingstudier. Tidsskrift for Norsk Psykologforening, 50, 1089–1095.
Fosse, R., Joseph, J., & Richardson, K. (2015). A critical assessment of the equal-environment assumption of the twin method for schizophrenia. Front Psychiatry, 6, 62. doi: 10.3389/fpsyt.2015.00062
Franke, B., Stein, J. L., Ripke, S., Anttila, V., Hibar, D. P., van Hulzen, K. J., … Sullivan, P. F. (2016). Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat Neurosci, 19(3), 420–431. doi: 10.1038/nn.4228
Fusar-Poli, P., & Meyer-Lindenberg, A. (2016). Forty years of structural imaging in psychosis: promises and truth. Acta Psychiatr Scand, 134(3), 207–224. doi: 10.1111/acps.12619
Gale, C. R., Hagenaars, S. P., Davies, G., Hill, W. D., Liewald, D. C., Cullen, B., … Harris, S. E. (2016). Pleiotropy between neuroticism and physical and mental health: findings from 108 038 men and women in UK Biobank. Transl Psychiatry, 6, e791. doi: 10.1038/tp.2016.56
Galhardo, R. S., Hastings, P. J., & Rosenberg, S. M. (2007). Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol, 42(5), 399–435. doi: 10.1080/10409230701648502
Gottesman, II, & Shields, J. (1967). A polygenic theory of schizophrenia. Proc Natl Acad Sci U S A, 58(1), 199–205.
Gottesman, I. (1991). Schizophrenia Genesis. New York: W.H. Freeman.
Greenwood, T. A., Lazzeroni, L. C., Calkins, M. E., Freedman, R., Green, M. F., Gur, R. E., … Braff, D. L. (2016). Genetic assessment of additional endophenotypes from the Consortium on the Genetics of Schizophrenia Family Study. Schizophr Res, 170(1), 30–40. doi: 10.1016/j.schres.2015.11.008
Gurung, R., & Prata, D. P. (2015). What is the impact of genome-wide supported risk variants for schizophrenia and bipolar disorder on brain structure and function? A systematic review. Psychol Med, 45(12), 2461–2480. doi: 10.1017/S0033291715000537
Hatzimanolis, A., Bhatnagar, P., Moes, A., Wang, R., Roussos, P., Bitsios, P., … Avramopoulos, D. (2015). Common genetic variation and schizophrenia polygenic risk influence neurocognitive performance in young adulthood. Am J Med Genet B Neuropsychiatr Genet, 168B(5), 392–401. doi: 10.1002/ajmg.b.32323
Hubbard, L., Tansey, K. E., Rai, D., Jones, P., Ripke, S., Chambert, K. D., … Zammit, S. (2016). Evidence of Common Genetic Overlap Between Schizophrenia and Cognition. Schizophr Bull, 42(3), 832–842. doi: 10.1093/schbul/sbv168
Inoue, K., Natsuyama, T., & Miyaoka, H. (2014). Case report of schizophrenia in adolescent with Russell–Silver syndrome. Psychiatry Clin Neurosci, 68(7), 582–582. doi: 10.1111/pcn.12169
Insel, T. R., & Cuthbert, B. N. (2009). Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry, 66(11), 988–989. doi: 10.1016/j.biopsych.2009.10.008
International Schizophrenia Consortium. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460(7256), 748–752. doi: 10.1038/nature08185
Jonas, R. K., Montojo, C. A., & Bearden, C. E. (2014). The 22q11.2 Deletion Syndrome as a Window into Complex Neuropsychiatric Disorders Over the Lifespan. Biol Psychiatry, 75(5), 351–360. doi: http://dx.doi.org/10.1016/j.biopsych.2013.07.019
Jones, H. J., Stergiakouli, E., Tansey, K. E., Hubbard, L., Heron, J., Cannon, M., … Zammit, S. (2016). Phenotypic Manifestation of Genetic Risk for Schizophrenia During Adolescence in the General Population. JAMA Psychiatry, 73(3), 221–228. doi: 10.1001/jamapsychiatry.2015.3058
Joseph, J. (2006). The missing gene: psychiatry, heredity, and the fruitless search for genes. New York: Algora.
Joseph, J. (2015). The trouble with twin studies: A reassessment of twin research in the social and behavioral sciences. New York: Routledge.
Kauppi, K., Westlye, L. T., Tesli, M., Bettella, F., Brandt, C. L., Mattingsdal, M., … Andreassen, O. A. (2015). Polygenic risk for schizophrenia associated with working memory-related prefrontal brain activation in patients with schizophrenia and healthy controls. Schizophr Bull, 41(3), 736–743. doi: 10.1093/schbul/sbu152
Kendler, K. S., & O’Donovan, M. C. (2014). A breakthrough in schizophrenia genetics. JAMA Psychiatry, 71(12), 1319–1320. doi: 10.1001/jamapsychiatry.2014.1776
Kundakovic, M., & Champagne, F. A. (2015). Early-life experience, epigenetics, and the developing brain. Neuropsychopharmacology, 40(1), 141–153. doi: 10.1038/npp.2014.140
Lancaster, T. M., Ihssen, N., Brindley, L. M., Tansey, K. E., Mantripragada, K., O’Donovan, M. C., … Linden, D. E. (2016). Associations between polygenic risk for schizophrenia and brain function during probabilistic learning in healthy individuals. Hum Brain Mapp, 37(2), 491–500. doi: 10.1002/hbm.23044
Lencz, T., Knowles, E., Davies, G., Guha, S., Liewald, D. C., Starr, J. M., … Malhotra, A. K. (2014). Molecular genetic evidence for overlap between general cognitive ability and risk for schizophrenia: a report from the Cognitive Genomics consorTium (COGENT). Mol Psychiatry, 19(2), 168–174. doi: 10.1038/mp.2013.166
Lewontin, R. C., Rose, S., & Kamin, L. J. (1984). Not in our genes: Biology, ideology and human nature. New York: Pantheon.
Liebers, D. T., Pirooznia, M., Seiffudin, F., Musliner, K. L., Zandi, P. P., & Goes, F. S. (2016). Polygenic Risk of Schizophrenia and Cognition in a Population-Based Survey of Older Adults. Schizophr Bull, 42(4), 984–991. doi: 10.1093/schbul/sbw001
Liu, B., Zhang, X., Cui, Y., Qin, W., Tao, Y., Li, J., … Jiang, T. (2016). Polygenic Risk for Schizophrenia Influences Cortical Gyrification in 2 Independent General Populations. Schizophr Bull. doi: 10.1093/schbul/sbw051
Maharjan, R., & Ferenci, T. (2015). Mutational signatures indicative of environmental stress in bacteria. Mol Biol Evol, 32(2), 380–391. doi: 10.1093/molbev/msu306
Malhotra, D., McCarthy, S., Michaelson, J. J., Vacic, V., Burdick, K. E., Yoon, S., … Sebat, J. (2011). High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron, 72(6), 951–963. doi: 10.1016/j.neuron.2011.11.007
Malhotra, D., & Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell, 148(6), 1223–1241. doi: 10.1016/j.cell.2012.02.039
Mark, W., & Toulopoulou, T. (2016). Cognitive intermediate phenotype and genetic risk for psychosis. Curr Opin Neurobiol, 36, 23–30. doi: 10.1016/j.conb.2015.08.008
Marshall, C., Howrigan, D., Merico, D., Thiruvahindrapuram, B., Wu, W., Greer, D., … Sebat, J. (2016). A contribution of novel CNVs to schizophrenia from a genome-wide study of 41,321 subjects. bioRxiv. doi: 10.1101/040493
McEwen, B. S., Bowles, N. P., Gray, J. D., Hill, M. N., Hunter, R. G., Karatsoreos, I. N., & Nasca, C. (2015). Mechanisms of stress in the brain. Nat Neurosci, 18(10), 1353–1363. doi: 10.1038/nn.4086
McIntosh, A. M., Gow, A., Luciano, M., Davies, G., Liewald, D. C., Harris, S. E., … Deary, I. J. (2013). Polygenic risk for schizophrenia is associated with cognitive change between childhood and old age. Biol Psychiatry, 73(10), 938–943. doi: 10.1016/j.biopsych.2013.01.011
Melis, J. P., Kuiper, R. V., Zwart, E., Robinson, J., Pennings, J. L., van Oostrom, C. T., … van Steeg, H. (2013). Slow accumulation of mutations in Xpc-/- mice upon induction of oxidative stress. DNA Repair (Amst), 12(12), 1081–1086. doi: 10.1016/j.dnarep.2013.08.019
Mulle, J. G., Pulver, A. E., McGrath, J. A., Wolyniec, P. S., Dodd, A. F., Cutler, D. J., … Conrad, D. F. (2013). Reciprocal Duplication of the Williams-Beuren Syndrome Deletion on Chromosome 7q11. 23 Is Associated with Schizophrenia. Biol Psychiatry.
Oertel-Knochel, V., Lancaster, T. M., Knochel, C., Stablein, M., Storchak, H., Reinke, B., … Linden, D. E. (2015). Schizophrenia risk variants modulate white matter volume across the psychosis spectrum: evidence from two independent cohorts. Neuroimage Clin, 7, 764–770. doi: 10.1016/j.nicl.2015.03.005
Papiol, S., Mitjans, M., Assogna, F., Piras, F., Hammer, C., Caltagirone, C., … Spalletta, G. (2014). Polygenic determinants of white matter volume derived from GWAS lack reproducibility in a replicate sample. Transl Psychiatry, 4, e362. doi: 10.1038/tp.2013.126
Pfeifer, G. P. (2006). Mutagenesis at methylated CpG sequences. Curr Top Microbiol Immunol, 301, 259–281.
Plaks, M., Argaman, R., Stawski, M., Qwiat, T., Polak, D., & Gothelf, D. (2010). Social-sexual education in adolescents with behavioral neurogenetic syndromes. Isr J Psychiatry Relat Sci, 47(2), 118–124.
Power, R. A., Steinberg, S., Bjornsdottir, G., Rietveld, C. A., Abdellaoui, A., Nivard, M. M., … Stefansson, K. (2015). Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat Neurosci, 18(7), 953–955. doi: 10.1038/nn.4040
Read, J., Bentall, R. P., & Fosse, R. (2009). Time to abandon the bio-bio-bio model of psychosis: Exploring the epigenetic and psychological mechanisms by which adverse life events lead to psychotic symptoms. Epidemiol Psichiatr Soc, 18(4), 299–310.
Read, J., Fosse, R., Moskowitz, A., & Perry, B. D. (2014). The traumagenic neurodevelopmental model of psychosis revisited. Neuropsychiatry, 4(1), 65–79.
Rees, E., Walters, J. T. R., Georgieva, L., Isles, A. R., Chambert, K. D., Richards, A. L., … Kirov, G. (2014). Analysis of copy number variations at 15 schizophrenia-associated loci. The British Journal of Psychiatry, 204(2), 108–114.
Rosenberg, S. M., Shee, C., Frisch, R. L., & Hastings, P. J. (2012). Stress-induced mutation via DNA breaks in Escherichia coli: a molecular mechanism with implications for evolution and medicine. Bioessays, 34(10), 885–892. doi: 10.1002/bies.201200050
Sanders, A. R., Duan, J., Levinson, D. F., Shi, J., He, D., Hou, C., … Gejman, P. V. (2008). No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am J Psychiatry, 165(4), 497–506. doi: 10.1176/appi.ajp.2007.07101573
Schizophrenia Working Group of the Psychiatric Genomic Consortium. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat Genet, 43(10), 969–976. doi: 10.1038/ng.940
Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511(7510), 421–427. doi: 10.1038/nature13595
Shevlin, M., Dorahy, M., & Adamson, G. (2007). Childhood traumas and hallucinations: an analysis of the National Comorbidity Survey. J Psychiatr Res, 41(3–4), 222–228. doi: 10.1016/j.jpsychires.2006.03.004
Shprintzen, R. J. (2008). Velo-cardio-facial syndrome: 30 Years of study. Dev Disabil Res Rev, 14(1), 3–10.
Sieradzka, D., Power, R. A., Freeman, D., Cardno, A. G., McGuire, P., Plomin, R., … Ronald, A. (2014). Are genetic risk factors for psychosis also associated with dimension-specific psychotic experiences in adolescence? PLoS One, 9(4), e94398. doi: 10.1371/journal.pone.0094398
Stankiewicz, P., & Lupski, J. R. (2010). Structural variation in the human genome and its role in disease. Annu Rev Med, 61, 437–455. doi: 10.1146/annurev-med-100708–204735
Stefansson, H., Meyer-Lindenberg, A., Steinberg, S., Magnusdottir, B., Morgen, K., Arnarsdottir, S., … Stefansson, K. (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature, 505(7483), 361–366. doi: 10.1038/nature12818
Stefansson, H., Ophoff, R. A., Steinberg, S., Andreassen, O. A., Cichon, S., Rujescu, D., … Collier, D. A. (2009). Common variants conferring risk of schizophrenia. Nature, 460(7256), 744–747. doi: 10.1038/nature08186
Sullivan, P. F., Kendler, K. S., & Neale, M. C. (2003). Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry, 60(12), 1187–1192. doi: 10.1001/archpsyc.60.12.1187
Terwisscha van Scheltinga, A. F., Bakker, S. C., van Haren, N. E., Derks, E. M., Buizer-Voskamp, J. E., Boos, H. B., … Psychiatric Genome-wide Association Study, C. (2013). Genetic schizophrenia risk variants jointly modulate total brain and white matter volume. Biol Psychiatry, 73(6), 525–531. doi: 10.1016/j.biopsych.2012.08.017
Terwisscha van Scheltinga, A. F., Bakker, S. C., van Haren, N. E., Derks, E. M., Buizer-Voskamp, J. E., Cahn, W., … Kahn, R. S. (2013). Schizophrenia genetic variants are not associated with intelligence. Psychol Med, 43(12), 2563–2570. doi: 10.1017/S0033291713000196
Van der Auwera, S., Wittfeld, K., Homuth, G., Teumer, A., Hegenscheid, K., & Grabe, H. J. (2015). No association between polygenic risk for schizophrenia and brain volume in the general population. Biol Psychiatry, 78(11), e41–42. doi: 10.1016/j.biopsych.2015.02.038
Varese, F., Smeets, F., Drukker, M., Lieverse, R., Lataster, T., Viechtbauer, W., … Bentall, R. P. (2012). Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective- and cross-sectional cohort studies. Schizophr Bull, 38(4), 661–671. doi: 10.1093/schbul/sbs050
Voineskos, A. N., Felsky, D., Wheeler, A. L., Rotenberg, D. J., Levesque, M., Patel, S., … Malhotra, A. K. (2016). Limited Evidence for Association of Genome-Wide Schizophrenia Risk Variants on Cortical Neuroimaging Phenotypes. Schizophr Bull, 42(4), 1027–1036. doi: 10.1093/schbul/sbv180
Walton, E., Hass, J., Liu, J., Roffman, J. L., Bernardoni, F., Roessner, V., … Ehrlich, S. (2016). Correspondence of DNA Methylation Between Blood and Brain Tissue and Its Application to Schizophrenia Research. Schizophr Bull, 42(2), 406–414. doi: 10.1093/schbul/sbv074
Walton, E., Turner, J., Gollub, R. L., Manoach, D. S., Yendiki, A., Ho, B. C., … Ehrlich, S. (2013). Cumulative genetic risk and prefrontal activity in patients with schizophrenia. Schizophr Bull, 39(3), 703–711. doi: 10.1093/schbul/sbr190
Weaver, I. C. (2009). Shaping adult phenotypes through early life environments. Birth Defects Res C Embryo Today, 87(4), 314–326. doi: 10.1002/bdrc.20164
Wei, W. H., Hemani, G., & Haley, C. S. (2014). Detecting epistasis in human complex traits. Nat Rev Genet, 15(11), 722–733. doi: 10.1038/nrg3747
Wray, N. R., Lee, S. H., Mehta, D., Vinkhuyzen, A. A., Dudbridge, F., & Middeldorp, C. M. (2014). Research review: Polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry, 55(10), 1068–1087. doi: 10.1111/jcpp.12295
Yeo, R. A., Gangestad, S. W., Walton, E., Ehrlich, S., Pommy, J., Turner, J. A., … Calhoun, V. D. (2014). Genetic influences on cognitive endophenotypes in schizophrenia. Schizophr Res, 156(1), 71–75. doi: 10.1016/j.schres.2014.03.022
Zammit, S., Hamshere, M., Dwyer, S., Georgiva, L., Timpson, N., Moskvina, V., … O’Donovan, M. C. (2014). A population-based study of genetic variation and psychotic experiences in adolescents. Schizophr Bull, 40(6), 1254–1262. doi: 10.1093/schbul/sbt146