Normal og patologisk kognitiv aldring - kan de skilles?
Knut Hestad & Ivar Reinvang
- Knut Hestad
- Ivar Reinvang
The present review discusses approaches to understanding factors behind the transition from normal to pathological aging and consequences for clinical neuropsychological assessment. Normal cognitive changes and dementia of Alzheimer’s type and of vascular origins are described. In the «normal» aging process there is a decline in many cognitive domains but quantitatively and qualitatively different from the two mentioned types of dementia. Alzheimer’s disease is a neurodegenerative disease with progressive loss of neurotransmitters that lead to learning and memory deficits and other cognitive problems. It is associated with increased accumulation of extracellular amyloid plaques and neurofibrillary tangles in the aging brain. There is a relationship to vascular dementia in that both disorders share common risk factors. In between normal and pathological aging the term «mild cognitive impairment» (MCI) has been introduced.
Keywords: normal aging, Alzheimer’s disease, vascular dementia, mild cognitive impairment, neuropsychological assessment.
Mange av risikofaktorene for Alzheimers sykdom og vaskulær demens er de samme. «Normal» aldring fører også med seg kognitiv svikt. Mellom patologisk utvikling og det «normale» benyttes gjerne termen mild kognitiv svikt.
Alder er et kronometrisk mål på tid som er gått. Hvis vi skal knytte et slikt tidsmål til nevropsykologiske funksjoner og endringer, så må alder først gis et innhold i form av biologiske og nevrologiske prosesser som kan gi grunnlag for å forstå endringene. Det har i senere år tilkommet en massiv strøm av ny viten som gjør det mulig i dag å se på den raskt økende levealder i den vestlige befolkning som et naturlig eksperiment der nevropsykologiske studier av relasjonen hjerne-atferd blir en sentral innfallsport til forståelse av aldersrelaterte kognitive endringer. Resultatene av slike studier kan også gjøre oss i stand til bedre å skille mellom normal og patologisk aldring. Dette skillet forskyver seg imidlertid etter hvert som kunnskapen øker, og etter hvert som muligheten for å forebygge eller behandle demenssykdommer i tidlige utviklingsstadier øker. En oppsummering av status og konsekvensene for nevropsykologisk praksis er hovedtema for denne artikkelen.
Normal aldring - biologiske og anatomiske endringerHjernebark
Det har lenge vært klart at hjernen endrer form og volum som følge av aldringsprosessen. Obduksjonsmaterialer viser at hjernens vekt reduseres og hulrommene blir større i eldre år. I de seneste årene har det vært mulig å kvantifisere disse endringene ved å studere MR-bilder av hjernen og foreta enten manuelle (ved å tegne omriss) eller automatiserte målinger gjennom datastyrt bildeanalyse. I norsk sammenheng er det særlig Anders Fjell og Kristine Walhovd (Fjell et al., 2005; Walhovd et al., 2005) som har anvendt avanserte metoder for bildeanalyse som har vært tilgjengelige gjennom et nært samarbeid med amerikanske miljøer. Slike undersøkelser viser at ulike områder av hjernebarken endrer seg i ulik takt med alderen. I noen områder er tykkelsen på hjernebarken nokså konstant, i andre områder er det en gradvis og rettlinjet fortynning i hele det voksne aldersspennet, og endelig er det noen områder som er stabile til en viss alder, men så viser akselererende fortynning. Foreløpig er det tverrsnittsdata som viser disse tendensene, og det vil ta tid før man også får gode longitudinelle data.
Følgende figur fra Walhovd et al. (2005) illustrerer de to nevnte aldersmønstrene.
Når en går nærmere inn på de ulike regioner av hjernebarken, så viser det seg noe overraskende at endringsmønstre kan være ganske lokaliserte, og at naboområder noen centimeter fra hverandre viser ulike mønster. Funnene bekrefter tidligere funn, at frontal korteks sannsynligvis viser fortynning med aldring, men ulike deler av frontallappen oppfører seg ulikt, og andre områder, for eksempel sensorisk-motorisk korteks, viser like store endringer som utvalgte frontale områder.
Fiberforbindelser
Alle komplekse kognitive funksjoner er avhengige av integrasjon av nevronal aktivitet, som både må evne å binde sammen områder fjernt fra hverandre og gjøre det med høy grad av presisjon. Hvordan dette fysiologisk sett er mulig, er fortsatt lite kjent, men studier av elektrisk hjerneaktivitet (EEG, ERP) har begynt å gi ledetråder. Uansett må de fibrene som forbinder ulike hjerneområder, være av høy kvalitet og i stand til å lede elektriske impulser raskt og presist. Myelinisering gir en isolerende skjede som er viktig for at nervefibre skal fungere godt, og studier av aldrende menneskeaper (Peters & Sethares, 2002,2003) har vist at myelinskjedene skades ved aldring. Myelin dannes av gliaceller, ikke av nevronene selv, og er i stand til å nydannes gjennom hele livet. Bartzokis (2004) mener at myelindannelse fortsetter opp til omkring 50 års alder, men at sent myeliniserende fibre er mer sårbare for skader enn tidlig myeliniserte. Konsekvenser av redusert myelinkvalitet er ineffektiv impulsoverføring og lekkasje av celler gjennom et skadet myelinlag. Ved hjelp av MR-måling av diffusjon («lekkasje») kan man finne relevante mål på fiberkvalitet. Corpus callosum består av myeliniserte fibre som forbinder symmetriske områder av hjernehalvdelene, og målinger der man har fokusert på fremre og bakre del av corpus callosum, viser at med aldring er fiberkvaliteten i fremre deler mer forringet enn fiberkvaliteten i bakre deler (Head et al., 2006). Slike funn er grunnlag for en økt interesse for endringer i hvit substans, som er viktige både i normal aldring og patologi.
Celleforandringer
Hvis vi så vil vite hva som skjer på cellenivå når hjernebark og hvit substans endres, så er det første spørsmålet i hvilken grad det skjer et massivt nevrontap med aldring. Studier av dødt hjernevev under mikroskop tydet i mange år på at det forelå et massivt nevrontap, inntil man fra midten av 1980-tallet og utover kunne påvise at det meste av funnene skyldtes de prepareringsprosedyrene som ble brukt. En oversiktsartikkel (Peters et al., 1998) konkluderer med at det ikke er noen entydig evidens for aldersrelatert kortikalt nevrontap. Derimot synes det klart at antall synapser og dendritter reduseres i «gamle» nevroner slik at nevronal plastisitet totalt sett er redusert. Det er en aldersrelatert opphopning av abnorme proteindannelser både i nevronene (tau-protein) og rundt nevronene (beta-amyloid). Disse er karakteristiske for Alzheimers sykdom, men er også utbredt i hjernen hos ikke-demente personer. Blodsirkulasjon i de finere forgreninger av årenettet svekkes og gir opphav til endringer som ut fra MR-bilder er karakterisert som inkomplette infarkter (O’Brien et al., 2003) uten at de dermed fører til karakteristiske symptomer på hjerneslag. På cellenivå og molekylært nivå er det økning i skader på grunn av oksidativt stress, redusert produksjon av signalstoffer og økende grad av cellulære feilfunksjoner (Mattson & Magnus, 2006). Resultatet er en opphopning av skadde molekyler og skadd DNA i cellekjernen av nevroner, men forfatterne konkluderer likevel med at «successfull neural ageing is possible for most people».
Hjerneaktivering
Konsekvenser av nevrobiologiske endringer kan påvises gjennom hjerne-aktiveringstudier med bruk av enten funksjonell MRI (fMRI) eller event-related potentials (ERP). Funnene tyder på at det er fysiologiske endringer i funksjon når aktiveringen er knyttet til enkle nevropsykologiske oppgaver. Oppmerksomhet er avhengig av raske prosesser for å flytte oppmerksomhetsfokus og for å filtrere ut distra-herende informasjon, og ERP egner seg godt til å studere oppmerksomhet på grunn av god tidsoppløsning (Reinvang, 1999). Resultatene er sammensatte, men tyder blant annet på at med aldring er det en mindre effektiv mekanisme for å registrere uventede endringer i omgivelsene. Dette viser seg både i forhold til automatisk og ubevisst registrering av endring, og i forhold til bevisst registrering (Cooper et al., 2006, Gaeta et al., 1998). Undersøkelser av hukommelse med fMRI har hovedsakelig vist at eldre viser et bredt sammensatt aktiveringsmønster, mens yngre viser et mer fokalt aktiveringsmønster i forhold til samme oppgave. Særlig gjelder dette under innkoding av ny informasjon, der eldre i hovedsak viser aktivering av begge frontallapper, mens yngre bare aktiverer venstre. Eldre som viser samme type aktiveringsmønster som yngre, presterer dårligere på oppgaven (Cabeza et al., 2002). Redusert spesialisering (dedifferensiering) eller aktivering av kompenserende mekanismer har vært diskutert som mulige forklaringer på endret hjerneaktivitet i eldre år.
Normal aldring - kognitive og nevropsykologiske endringer
Aldringseffekten på kognitive funksjoner kan avleses mer eller mindre direkte i normene for nevropsykologiske tester. Hestad (1998) har oppsummert de kjente forskjellene i aldersforandring mellom flytende og krystallisert intelligens slik de kommer fram på Wechsler-testene og andre oppgaver som krever henholdsvis problemløsning og mer kunnskapsbaserte funksjoner. En mer detaljert inndeling vil ofte fokusere på hukommelse, eksekutive funksjoner og oppmerksomhet der kognitive aldringseffekter er godt kartlagt.
Hukommelse
Hukommelse ses i kognitiv psykologi som sammensatt av subfunksjoner, og en stort anlagt studie av hukommelse og aldring er BETULA-studien fra Sverige der aldersgruppen fra 35 til 80 er studert med data både fra tverrsnittsundersøkelser og longitudinelle undersøkelser (Nilsson et al., 1997). Resultatene viser at episodisk hukommelse målt med hukommelse for navn, ordlister, historier, ansikter og annet viser en jevn reduksjon gjennom hele det studerte aldersspennet. Park et al. (2002) finner at samme tendensen gjelder helt ned til 20 års alder og både for verbalt og visuo-spatialt materiale. Parallelt med en reduksjon i hukommelse for episodisk informasjon er det en økning i tendensen til å produsere falske minner med økende alder. Det ses blant annet på gjenkjenningsoppgaver der det inngår nye ord eller bilder som er assosiert med tidligere presentert materiale, og på oppgaver der man i tillegg til å huske et ord må huske sammenhengen ordet ble presentert i (Koutstaal & Schacter, 1997; Spencer & Raz, 1995). Chua, Chen & Park (2006) undersøkte om hukommelse for kontekstuell informasjon svekkes mindre med alderen i Kina, en kultur som legger større vekt på kontekstuell forståelse, men fant ingen forskjell i aldringseffekt. Semantisk hukommelse målt med tester på ordforråd og kunnskaper viser økning fram til nærmere 60 år, deretter reduksjon. Resultatene for semantisk hukommelse er dårligere dersom det er tidspress på ordframkalling, som ved ordflytoppgaver, som går ut på å nevne så mange ord fra en gitt kategori som mulig på begrenset tid. Implisitt hukommelse ser ikke ut til å vise markerte aldersendringer.
Oppmerksomhet
Oppmerksomhet er nevnt ovenfor i forbindelse med elektrofysiologiske studier av basale mekanismer. På nevropsykologisk plan er en allmenn reduksjon i tempo med aldring et gjennomgående fenomen som ses i alle tester der tidsmål inngår, fra enkle motoriske tester til sammensatte problemløsningsoppgaver. Denne effekten er såpass gjennomgripende og global at den vanskelig lar seg tolke som reduksjon i noen spesifikk nevropsykologisk funksjon. Et optimistisk perspektiv er at forskjellene mellom yngre og eldre kan reduseres når eldre får øve på oppgaven (Kramer & Kray, 2006). Studier som har lett etter mer spesifikke alderseffekter på oppmerksomhet, fokuserer på selektive effekter som ikke lar seg forklare med generell langsomhet. Det er ulike måter å inndele oppmerksomhetsfunksjoner på, men de har det til felles at alle skiller mellom en automatisk styring av oppmerksomhet og en kontrollert styring. Kramer & Kray (2006) har oppsummert studier av aldring og oppmerksomhet og finner ganske store forskjeller mellom effekten av aldring i ulike oppmerksomhetskrevende situasjoner. De finner at oppgaver som krever oppmerksomhet for flere elementer samtidig og veksling mellom dem, viser markert reduksjon med alderen. Slike oppgaver krever en form for kontrollert eller eksekutiv oppmerksomhet som antagelig er avhengig av frontallappsfunksjoner. På den annen side finner Parasuraman & Greenwood (1998) at en oppgave der visuell oppmerksomhet styres mot ulike deler av omgivelsene av signaler («cues»), bare viser små alderseffekter. Disse oppgavene er antagelig mer avhengige av intakte parietallappsfunksjoner, som i større grad er automatiserte.
Eksekutive funksjoner
Eksekutive funksjoner er et sammensatt begrep som omfatter planlegging og problemløsing, men som også brukes som en fellesnevner for funksjoner som kan assosieres med funksjon i prefrontale hjerneområder. Noen vil også oppfatte integrasjon mellom kognitive og emosjonelle funksjoner som en sentral del av eksekutive funksjoner (Tranel, 2002). Nevropsykologiske oppgaver som anses som mål på eksekutiv funksjon, omfatter Wisconsin Card Sorting Test, Tower of Hanoi, Stroop test, Trail Making test B og andre. Alle disse viser klare aldersrelaterte endringer, og funnene har blitt tatt til støtte for at frontallappsforandringer er sentrale for kognitiv aldring (West, 1996). Selv om selektive utfall på disse testene kan gi holdepunkter for frontale hjerneskader, så er det mer usikkert hva de måler i en normal aldringssammenheng. Arbeidsminne («working memory») er etter manges oppfatning en sentral komponent i oppgaver som stiller store krav til eksekutive funksjoner. Eksekutivt arbeidsminne innebærer å holde informasjon aktiv og i sentrum for oppmerksomheten mens man også manipulerer informasjonen. Kjente nevropsykologiske oppgaver som Tallspenn Baklengs, Paced Auditory Serial Addition test, Bokstav-tall-sekvensering og Trail Making B fanger opp disse momentene, og det er sannsynlig at også komplekse problemløsningsoppgaver som Wisconsin Card Sorting er sterkt avhengige av velfungerende arbeidshukommelse. Arbeidshukommelse viser en klar lineær svekkelse med økende alder fra ung voksen alder (20 år) oppover til 80 år (Park & Payer 2006). Det er en rimelig antagelse at reduksjon i den eksekutive delen av arbeidshukommelse forklarer en stor del av de aldersbetingede endringene i andre eksekutive funksjonsmål. En pågående diskusjon i litteraturen handler om hvorvidt problemet med arbeidshukommelse skyldes redusert kapasitet, økt interferens eller andre faktorer (Paxton et al., 2007; Park & Payer, 2006).
Forklaringsmodeller
Alderseffekter på kognisjon kommer altså fram både innenfor hukommelse, oppmerksomhet og eksekutive funksjoner. Det er i seg selv ikke overraskende siden de basale nevrobiologiske forandringene ved aldring (se ovenfor) er bredspektrede og må antas å påvirke et tilsvarende bredt spekter av funksjoner. Det er likevel mulig å spørre om de kognitive forandringene kan skyldes endring i et lite antall underliggende kognitive faktorer som så slår ut på flere områder. Den mest utbredte teorien har vært den som er lansert av Salthouse (1996), som sier at prosesseringshastighet («processing speed») er en faktor som forklarer en stor del eller all variansen i kognitive aldringsdata. Selv om dette langt på vei kan være riktig ut fra et statistisk modelleringssynspunkt, så gir det ikke god mening fra et nevropsykologisk synspunkt, der vi ser på hjernen som sammensatt av spesialiserte funksjoner. Gjennomgangen ovenfor tyder også på at selv om komplekse psykometriske tester viser globale aldersforandringer, så viser undersøkelser med eksperimentelle kognitive paradigmer et mer differensiert mønster. Craik og Bialystok (2006) har et mer nyansert syn på kognitiv aldring, der de gjør et grunnleggende skille mellom representasjonsfunksjoner og kontrollfunksjoner. Representasjonsfunksjoner er relativt stabile med aldring, og en større integrasjon mellom kunnskap og kognitive strategier gir en økning av ekspertkompetanse med alder. Ekspertkompetanse kan langt på vei opprettholdes selv når kontrollfunksjoner svekkes. Kontrollfunksjoner svekkes med alder fra tidlig voksen alder, og eksekutiv arbeidshukommelse og sammensatt oppmerksomhet er mest rammet.
Variasjon i aldringsprosessen
Både de biologiske og de nevropsykologiske dataene viser generelle trekk som gir verdifull innsikt i aldringsmekanismer og deres konsekvenser. Begge typer data viser imidlertid også at det er store variasjoner innenfor hver aldersgruppe (noen 70-åringers hjerner ligner mer på 30-åringer enn på sine jevnaldrende, se figur 1). Det er kjent at tverrsnittsundersøkelser (sammenligning mellom aldergrupper) viser større aldringseffekter enn longitudinelle undersøkelser (oppfølgning av samme personer over tid). Begge typer undersøkelser har sine svakheter. Det viktigste er å understreke at det er betydelig individuell variasjon i aldringsforløpet, og at både miljømessige (aktivitet, kosthold) og genetiske faktorer påvirker denne variasjonen. Et mål for helsemyndigheter framover er å legge til rette for en sunn hjerne så langt inn i aldringsforløpet som mulig (Anderson & McConnell, 2007).
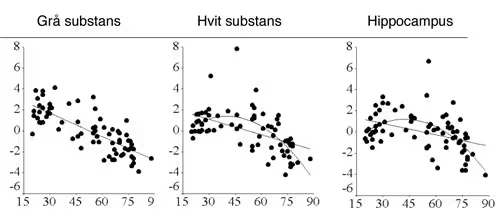
Figur 1. Sammenheng mellom alder og mengde hjernevev. Hjernevev målt med automatisert bildeanalyse av MR-bilder viser en lineær reduksjon i aldersspennet 20 til 90 år. For hvit substans og hippocampus er det også en tiltagende reduksjon etter ca. 60 år. Gjengitt etter Walhoved et al. (2005) med tillatelse fra forfatterne.
Demens
Ved aldring er det en rekke biologiske forhold og sykdommer som personen er mer sårbar for enn tidligere i livet, men demens er den lidelsen ved aldring som nevropsykologer kommer mest i kontakt med.
Demens er en lite spesifikk diagnose. Den gir en beskrivelse av atferdsmessig svikt som forsøkes relatert til hjerneskade, dvs. både strukturelle og molekylære skader. Ifølge ICD-10 (WHO, 1993) beskrives demens som en reduksjon i hukommelse, særlig ny informasjon, men i mer alvorligere tilfeller også gjenkalling av tidligere lært materiale. Dette gjelder både verbalt og ikke-verbalt materiale. Også andre kognitive funksjoner kan svikte, med språkforstyrrelser, eksekutive funksjoner, apraksi og agnosi. Svikten bør bli verifisert ved å oppta historie fra informant, supplert dersom mulig, ved nevropsykologiske tester eller kvantifisert ved kognitiv undersøkelse. Alvorlighetsgrad klassifiseres etter et sett av kriterier. Prevalensen av demens er ca 1 % ved 65 år og 50 % ved 90 år (Hoffman et al., 1991). Kvinner har en større andel demens enn menn, men her er det mange faktorer som spiller inn. Blant annet lever kvinner lenger enn menn. Imidlertid, selv om slike faktorer trekkes inn, er det allikevel en større risiko for kvinner enn for menn til å utvikle demens. DSM-IV legger vekt på at svikten må være så stor at den går ut over arbeid eller sosiale funksjoner, mens ICD-10 understreker at forandringen må ha vart over en viss tid.
De to dominerende årsakene til demens hos eldre erAlzheimers sykdom og cerebrovaskulære forandringer. Sistnevnte omfatter hjerneslag, småkarsvikt og iskemiske endringer. Førstnevnte er en degenerativ cerebral lidelse med påfølgende progredierende demens. Siden disse to demenslidelsene er såpass dominerende, begrenser vi vår omtale av demenslidelser til disse to diagnosene.
Alzheimers sykdom
Den mest vanlige form for demens er Alzheimers sykdom, som teller ca. 60 - 70 % av alle demenslidelser (Fratiglioni et al., 2000). Diagnosen Alzheimers sykdom ble første gang brukt av Emil Kraepelin etter at Alois Alzheimer beskrev en slik pasient i 1907. Ifølge ICD-10 finnes Alzheimers sykdom som nevrologisk diagnose (som kan forekomme uten demens) og demens ved Alzheimers sykdom.
Det er tre forskjellige systemer som brukes for å diagnostisere demens ved Alzheimers sykdom. ICD-10, DSM-4 og kriteriene utviklet av «The National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and Alzheimer’s disease and Related Disorders Association (ADRDA)» (McKhan et al., 1984). Ved å bruke NINCDS-ADRDA-kriteriene opererer man med begrepene sannsynlig (probable), mulig (possible) og definitiv (definite) demens av Alzheimers type. Alle systemene har med kriterier for svekkelse av kognitive funksjoner, særlig legges det vekt på hukommelse, men også andre kognitive funksjoner som afasi, tenkning, eksekutive funksjoner med mer. Forstyrrelsene må være så store at de går ut over dagliglivets funksjoner. En definitiv diagnose av Alzheimers sykdom krever histopatologisk bekreftelse ved obduksjon.
Biologisk grunnlag for utvikling av demens ved Alzheimers type
Alzheimer beskrev plakk og nevrofibrillære nøster, samt arteriesklerotiske forandringer i hjernen på sin pasient «Fru Auguste D». Nevrofibrillære nøster er forandringer som finner sted inne i selve nevronene. Normalt finnes fine proteinholdige fibere inne i cellen som forløper parallelt. Ved Alzheimers sykdom tvinner disse fibrene seg sammen og rundt hverandre i en såkalt alfa-heliks-struktur og danner nøster. Antagelig dør cellen når denne fibertvinningen når et maksimum. Hvorfor dette skjer, vet man ikke sikkert. Forandringene sees utbredt både i korteks, de limbiske strukturer i hjernen og i visse sentra i hjernestammen. I startfasen av lidelsen er det særlig områdene hippocampus (Andersson, 2007) og entorhinal korteks (Braak & Braak, 1991) som er affisert.
Senile plakk finner man i ytterkant av cellene knyttet til dendrittene. Disse forandringene kaller man gjerne ekstracellulære forandringer; de består av oppsamling av et protein, kalt beta (b)-amyloid, som ligger i sentrum av ødelagte nerveutløpere til flere nevroner. b-amyloidet dannes i hjernen og finnes først og fremst i de kortikale strukturene ved Alzheimers sykdom. Det er gjort funn av markører knyttet til både plakk [reduksjon av 42 aminosyreform av beta-amyloid (Ab42)] og nøster [øket total-tau (T-tau) og hyperfosforylert tau (P-tau)] i spinalvæske. Det er foreslått at forhøyet CSF P-tau er mer spesifikk for Alzheimers sykdom enn de andre to markørene, selv om det her er mye usikkerhet (Galasko et al., 1998; Blennow & Vanmechelen, 2005). Resultatet av disse to prosessene er at nevronet dør.
Det må imidlertid understrekes at både plakk og nøster finnes i hjernen hos «friske» eldre uten Alzheimers sykdom. Dersom man sammenlikner hjernen til henholdsvis en frisk eldre 85-åring med hjernen til en 85-åring med nyoppdaget Alzheimes sykdom, er det kun kvantiteten av plakk og nøster som er forskjellig, sannsynligvis ikke kvaliteten. Forskjellene i denne kvantiteten er imidlertid mye større om sammenlikningen gjøres hos Alzheimer-pasienter på 50 år og like gamle friske personer enn hos tilsvarende 85-åringer. Plakk og særlig nøster er heller ikke spesifikt for Alzheimers sykdom, de finnes også i flere andre hjerneorganiske sykdommer.
I tillegg til disse funnene, som på mange måter følger opp de opprinnelige funnene Alzheimer gjorde, er det sett endringer i blodtrykk knyttet til demens. Disse endringene kan relateres både til demens ved Alzheimers sykdom og vaskulær demens. Det er da også slik at mange som utvikler demens har en blanding eller overlapp av de to nevnte typer demens. Særlig er det sett at lavt blodtrykk heller enn høyt er relatert til demens hos de eldste (Hestad, Kveberg & Engedal, 2005). Populasjonsbaserte studier har konkludert med at midtlivs forhøyet blodtrykk kan være relatert til kognitiv svikt, lavt blodtrykk og demens sent i livet (Launer, Masaki, Petrovik, Foley & Havlik, 1995; Kilander, Nyman, Boberg, Hansson & Lithell, 1998; Swan, Carmelli & Laurue, 1998; Kivipelto, Helkala, Hänninen, Laakso, Hallikainen, Alhainen et al., 2001). Høyt blodtrykk foregriper Alzheimers sykdom med dekader og resulterer i en tidligere debut for demens ved Alzheimers sykdom enn den ellers ville gjøre (Skoog & Gustafson, 2003). Det lavere blodtrykket i høy alder kan således også være uttrykk for tilkommet hjerneskade.
Klinikk og nevropsykologi
Hukommelsesvansker er gjerne det første kognitive tegnet på at noe er galt. Slike problemer kommer ofte til syne på nevropsykologiske tester 2 - 3 år før pasienten får en klinisk diagnose (Andersson, Lindau, Almkvist et al., 2006). Den typiske pasienten med Alzheimers sykdom som er dement, kan fortsatt i noen grad lære hvordan handlinger utføres, men er ikke, eller i liten grad, i stand til å gjenta eller gjenkalle hva som skjedde for bare noen minutter siden. De kan klare å gjenkalle etter noen sekunder, men så er det vanskelig eller umulig å hente ting fra hukommelsen når det strekker seg over minutter eller timer.
Hestad, Dybing og Kløve (1997) undersøkte normalt friske eldre og pasienter som var henvist til en geriatrisk klinikk for hukommelsesvanskeligheter, med Hopkins Verbal Learning test (HVLT). Alzheimerpasientene, som i gjennomsnitt var 70 år gamle, hadde betydelige hukommelsesproblemer av forskjellig slag, men særlig var den utsatte gjenkallingen redusert. Det vil si at når det hadde gått noen minutter, så klarte ikke pasientene med Alzheimers sykdom å fritt gjenkalle ordlisten som tidligere var presentert for dem. Det var også vanskeligheter med selve innlæringen, men den utsatte hukommelsen viste seg å være det største problemet hos Alzheimer-pasientene. Et interessant moment ved Alzheimer-pasientene i ovennevnte undersøkelse, var at når de fikk holdepunkter til å hjelpe seg for å huske (dvs. gjenkjenning), viste det seg at ikke alt var tapt fra hukommelsen. Tvert imot, med holdepunkter klarte de å huske forbausende mange ord fra ordlisten. Dette tyder på at det er selve det å få ordene frem som kan være det vanskelige. Pasientene klarer altså å gjenkjenne, men ikke å gjenkalle. Dette er av betydning for disse pasientene, fordi det antagelig kan bety mye når det gjelder å benytte så mye som mulig av hukommelsen.
De samme hukommelsesvanskelighetene som ovenfor er beskrevet, gjelder også for gjenkalling av spatialt materiale, for eksempel det å gjenkjenne ansikter, finne frem i nye omgivelser osv. Hukommelsesproblemene til Alzheimer-pasientene vil øke på etter som sykdommen utvikler seg, slik at det blir vanskelig for pasienten å huske ting som har skjedd tidligere i livet. Dette har sannsynligvis sammenheng med at sykdommen rammer stadig større deler av hjernen med ødeleggelse av hjernebarken. Det er også relativt store variasjoner i startfasen for hvordan den kognitive svikten arter seg. Ved siden av hukommelsessvikt sees ofte på et tidlig tidspunkt vansker med språk og visuospatiale ferdigheter. Likeledes sees på et tidlig stadium i utviklingen svekkelse av høyere kognitive ferdigheter som abstraksjonsevne, fleksibilitet i tankegangen, såkalte eksekutive funksjoner og persepsjonevne.
Det er heller ikke uvanlig at pasienter med Alzheimers sykdom utvikler emosjonelle vanskeligheter med engstelse og/eller depresjonsliknende tilstander. Det har vært pekt på at depresjon sammen med demensutviklingen gir en dårligere prognose med hensyn til overlevelse enn bare demens alene. I omtrent 50 % av tilfellene er de første objektive symptomene ikke hukommelsesvansker, men psykiatriske eller andre nevrologiske vansker (Gustafson & Hagberg, 1975; Oppenheim, 1994).
Undergrupper
Det er foreslått at det finnes undergrupper av Alzheimers sykdom som kan vise et annet klinisk bilde enn det typiske, hvor fokus ikke i første omgang er knyttet til hippocampus (temporale-limbiske strukturer) og temporallappsområdet, dvs. det er ikke hukommelsesproblemene som dominerer. Det er vist at pasienter med Alzheimers sykdom både kan ha fokus knyttet til frontallappene og parietal-oksipitallappene (Furey-Kurkjian et al., 1995). Ved frontallappsskader sees oftere forandringer av personlighet, eksekutive funksjoner og språkvansker, mens ved skader i parietal-oksipitallappsområdet blir det problemer blant annet med synspersepsjon og visuo-spatial orientering. Spørsmålet om undergrupper forblir uavklart så lenge sikker diagnose av Alzheimers sykdom forutsetter tilgang til obduksjonsmateriale.
Arv
Som allerede nevnt har mange med Alzheimers sykdom en vesentlig arvelig belastning for sykdommen, men langt fra alle, og det er heller ingen klar arvegang. Det er funnet forskjeller i forhold til «det normale» når det gjelder arv med mutasjoner i kromosompar 1, 14, 19 og 21, som alle er funnet å ha betydning for Alzheimers sykdom. Når det gjelder 1,14 og 21, så er disse ansvarlige for familiær Alzheimers sykdom. For de som utvikler Alzheimers sykdom ved sen debut (etter 65 år), som er de fleste, er det polymorfisme knyttet til kromosompar 19 en særlig har vært opptatt av. Her er det apolipoproteingenet (Apo), som eksisterer i tre forskjellige former for alleler, som er av betydning: e2, e3, og e4. Ett gen fra far og ett fra mor. Det må understrekes at genet ikke bestemmer hvem som får Alzheimers sykdom, men kun er en risiko- eller sårbarhetsfaktor (Strittmatter et al., 1993). Slik sett er e4 av en helt annen natur enn hos de som har den familiære typen av Alzheimers sykdom, hvor risiko for sykdommen er langt større enn ved å ha ett eller to e4-alleler.
Vaskulær demens (VD)
Vaskulære cerebrale problemer er den nest vanligste årsaken til demens og teller mellom 10 - 50 % av alle tilfellene. Det er gegografiske variasjoner, forskjeller i pasientpopulasjon og klinisk tilnærming til diagnose som gjør at tallene varierer svært mye (Lobo, Launer, Fratiglioni et al., 2000). En stor utfordring ved VD er fravær av samlende retningslinjer for diagnose. Dette har blant annet sin bakgrunn i at evidens-baserte studier ofte mangler klare definisjoner på denne lidelsen. I 1992 foreslo en spesialgruppe diagnostiske kriterier for vaskulær demens (Román, Tatemichi, Erkinijuntti et al., 1993). Her konkluderes det med at vaskulær demens har en heterogen bakgrunn med hensyn til patologiske subtyper som inkluderer cerebrale infarkter og blødninger, hvor hypoksi kan være av betydning, samt senil (aldersmessig) ødeleggelse av hvit substans. Nesten alle typer hjerneslag (infarkter og/eller blødninger) vil kunne gi et symptombilde som faller inn under definisjonen demens, dvs. at den kognitive svikten blir så alvorlig at den representerer en betydelig svekkelse i sosial eller yrkesmessig fungering, med en betydelig reduksjon sammenlignet med tidligere funksjonsnivå (DSM-IV). Siste skudd på stammen av begreper relatert til kognitiv svikt ved cerebrovaskulære lidelser er «vascular cognitive impairment» (Erkinjuntti, 2007). Begrepet refererer til alle etiologier av cerebrovaskulære sykdommer inkludert vaskulære risikofaktorer som kan resultere i hjerneskade som igjen gir kognitiv svekkelse. Slik sett rommer begrepet mer enn kun vaskulær demens.
Hjerneslag-demens
Frekvens av demens etter hjerneslag varierer i nyere studier fra 12 - 32 % innefor 3 - 12 måneder etter slaget (Tatemichi, Desmond Mayeux, et al., 1992; Leys, Henon, Mackowiak-Cordoliani, Pasquier, 2005). Siden mange av VD-pasientene i utgangspunktet har hjerneslag, har det vært vesentlig å se den tidsmessige sammenhengen mellom slaget og demensutviklingen. Det er langt flere menn enn kvinner som rammes av VD, og prevalenstallene øker med økende alder.
Strukturelle endringer, hvor i hjernen
Vaskulær demens er altså ikke én type lidelse. På samme måte som hos slagpasienter betyr det mye hvor i hjernen og hva slags vaskulær skade det er, for hvordan det nevropsykologiske skadebildet blir. Gjennomsnittsdata og standard avvik kan derfor ved VD være nokså meningsløst. Det er få typer demens som har et så mangslungent bilde som vaskulær demens.
Når det gjelder hva slags cerebrovaskulære skader som er viktigst for demensutviklingen, er dataene sprikende. Det er imidlertid indikasjoner for at bilaterale iskemiske lesjoner heller enn unilaterale lesjoner er av betydning for demensutvikling (Erkinjuntti, Lee, Gao, Steenhuis, Eliasziw, Fry, Merskey, Hachinski, 1993; Erkinjuntti & Hachinski, 1993). Hvor i hjernen skadene fant sted, syntes også å være viktig, men hvilke lokalisasjoner som synes mest viktige, er det ingen klar enighet om. Noen forfattere poengterer dype infarkter i frontale og limbiske områder, andre i kortikale områder, særlig i temporale og parietale områder. Det er uenighet om antall og volum på infarktene. Hvit substans-forandring kan også være av betydning, idet det leder til funksjonelle brudd i forbindelsen mellom kortikale hjerneområder.
Erkinjuntti og Hachinski (1993) foreslo at spesielt kritiske områder for vaskulær demens var limbiske og paralimbiske områder, dienkefalon, basale forhjerne, frontallappene og tilliggende hvit substans. Disse områdene inkluderer forbindelser til hippocampus, parahippocampus, mediodorsal nukleus av thalamus, frontale områder og hvit substans rundt hodet av nucleus caudatus. Lesjoner i de ovennevnte områdene affiserer ny læring og mentale prosesser som omfatter sekvensering, planleggende og utøvende funksjoner samt selvvurderende og selvkritisk atferd.
Kliniske og nevropsykologiske endringer
Heterogeniteten ved vaskulær demens er stor, og variasjonene i patologiske lesjoner gjør at man kan klassifisere i forskjellige undergrupper (Murray, Knopman, Dickson, 2007).
Svært mange slagpasienter får hukommelsesproblemer i tillegg til andre kognitive svekkelser. Slagpasienter med skader i venstre hjernehemisfære får gjerne språkproblemer i større eller mindre grad, og svært mange med skader i venstre hjernehemisfære får også apraksi. Om skaden er i høyre hemisfære (bak optisk schiasma), blir det gjerne neglekt og synsfeltsutfall om synsnerven rammes. Er skaden fortil i hjernen, blir det ofte problemer med eksekutive funksjoner. Skadebildet like etter hjerneslaget er oftest så stort at det går ut over pasientens sosiale og yrkesmessige funksjoner. Det er derfor slik at den nevropsykologiske svikten må være av en viss varighet for å bli kalt demens. De fleste slagpasienter kommer seg betraktelig i den første tiden etter hjerneslaget. Det samme kan mange ganger sees hos pasienter som utvikler vaskulær demens som følge av hjerneslag. Demens har ofte blandede årsaker, men ofte er det holdepunkter for VD heller enn Alzheimers sykdom dersom pasienten viser symptomer med gangvansker, inkontinens og humør- eller personlighetsforandringer.
Sammenheng mellom demens ved Alzheimers sykdom og vaskulær demens
Cerebrovaskulære sykdommer kan forverre demens ved Alzheimers sykdom. I tillegg konkluderer mange studier med at cerebrovaskulære risikofaktorer er sterkt assosiert med Alzheimers sykdom (Wu, Munas, Petkov et al., 2002; Launer, Ross, Petrovitch et al., 2000; Kivipelto, Helkala, Hanninen, et al., 2001; Hestad & Engedal, 2006). Disse lidelsene overlapper hverandre (Anderson, 2007). Med økende alder viser pasienter med vaskulær demens økende akkumulering av b-amyloid sammenliknet med eldre uten cerebrovaskulær sykdom. Dette indikerer at pasienter med vaskulær demens får Alzheimer-liknende forandringer i høy alder (Lewis et al. 2006). Med andre ord er vaskulære faktorer assosiert med Alzheimer-forandringer i hjernen (Korf et al., 2005). Praktisk talt alle risikofaktorer for Alzheimers sykdom har en vaskulær komponent som minsker cerebral blodgjennomstrømning (de la Torre, 2002). Disse funnene underbygger at demens ved Alzheimers sykdom og vaskulær demens ikke er diagnoser som utelukker hverandre; de deler både vaskulære og nevrodegenerative patologiske sider (Lewis et al. 2006). Vaskulær demens og demens ved Alzheimers sykdom er i stor grad overlappende tilstander.
Behandling
Alzheimers sykdom er en progredierende lidelse som vi per dags dato ikke kan gjøre noe mer med enn i beste fall å bremse symptomutviklingen i en periode. Farmakologisk har det vært to tilnærminger som har gitt noe resultat: acetylkolinesterasehemmere (Rogers, Farlow, Doody, Mohs, Friedhoff, 1998) og N-metyl-D-asparate (NMDA) glutamatantagonister (Parsons, Stoffler, Danysz, 2007). De førstnevnte hemmer nedbrytingen av acetylkolin, som er av betydning blant annet for hukommelse, mens medisinen Memantine er en NMDA-reseptorkanalblokker. Her er poenget at hjernen igjen skal bli i balanse med hensyn til glutamat. Blokade av NMDA-reseptorer kan lede til reduksjon av nevronal plastisitet, noe som har betydning for læring og hukommelse (Collingridge & Bliss, 1995), mens overaktivitet leder til celledød på grunn av kalsiumbelastningen (Choi, 1992). Men samtidig er det slik at dysfunksjonell overaktivering av NMDA-reseptorer kan lede til forstyrret plastisitet (Parsons, Stoffler, Danysz, 2007). Her gjelder det da å skape den riktige balansen.
Resultatene fra begge typer medisiner er i beste fall at det skjer en viss bremsing av symptomutviklingen i en relativt kort periode. Den sistnevnte hører til de nyeste medisinene mot Alzheimers sykdom, selv om det lenge har vært kjent som legemiddel. Sannsynligvis er ikke effekten mye større for noen av dem enn hva miljøterapeutiske tiltak ville ha gitt.
Når det gjelder vaskulær demens, er det stort sett behandling som ved hjerneslag.Det gjelder å forebygge nye hjerneslag, eller nye cerebrovaskulære insidenser. På grunn av den fysiologiske beskaffenheten ved aldring er det en øket risiko for hjerneslag, både infarkter og blødninger som skaper nevropsykologiske utfall. Økt blodtrykk er en slik risiko. I en studie (Hestad, Kveberg & Engedal, 2005) av folk over 80 år ble det sett at over halvparten av deltagerne benyttet seg av blodtrykksreduserende medisiner. Samtidig ble det sett at det var lavt blodtrykk som korrelerte best med dårlig skåre og demens på Mini Mental State Examination (MMSE). Teorien her er at høyt blodtrykk tidligere i livet har gitt skade som både har redusert blodtrykket og gitt demensutvikling. Blodtrykket faller gjerne noe før demensutviklingen blir synlig (Skoog, 2003). Videre er det sett at blodtrykksreduserende medisiner kan være av betydning både for å forebygge demens (Hestad & Engedal, 2006) og Alzheimers sykdom (Guo et al., 2001), særlig synes det å være av betydning når pasienten har Apo E-e4-genet (Guo et al., 2001; Hestad & Engedal, 2006). Hva mekanismene bak dette er, er uklart, men det kan være av betydning at noen av de blodtrykksreduserende medisinene er kalsiumkanalantagonister og derved bremser influks av kalsium til cellene (Hestad & Engedal, 2006).
MCI - mild kognitiv svikt
Ved siden av demens er det kommet til et begrep mild kognitiv svikt (mild cognitive impairment, MCI) (Petersen, Smith, Waring et al., 1997). Det meste av arbeidet som her er gjort, retter seg mer mot demens ved Alzheimers sykdom enn mot vaskulær demens. En har derfor lagt vekt på svikt i hukommelsen som en risikofaktor atskilt fra andre kognitive variabler, og det er godt grunnlag for å hevde at nevropsykologisk dokumentert svikt i hukommelsen (amnestisk MCI) innebærer en klar økning i risiko for å utvikle Alzheimers sykdom (Petersen, 2004). Derfor er også denne typen kognitiv svikt tatt med i de forskningskriteriene for tidlig diagnostikk av Alzheimers sykdom som er foreslått av DuBois et al. (2007), men ikke som et tilstrekkelig kriterium for en diagnose. MCI kan også være karakterisert ved svikt i andre kognitive funksjoner enn hukommelse, og er en heterogen klinisk entitet. Det er ingen enighet om den kliniske betydningen av nevropsykologiske avvik i oppmerksomhet, eksekutive funksjoner eller andre funksjoner hos eldre. Når det er kognitiv svikt hvor ikke hukommelseproblemer dominerer, er det mer sannsynlig at årsaken er cerebrovaskulær sykdom enn Alzheimers sykdom (Graham, Rockwood, Beattie et al., 1997; Riley, Snowdon, Markesberry, 2002). Allikevel, ca. 30 % av pasienter med mild kognitiv svikt har hukommelsessvikt som er klinisk og patologisk forenlig med Alzheimer-patologi (Graham, Rockwood, Beattie et al., 1997; Riley, Snowdon, Markesberry, 2002; DeCarli, 2003). Når det gjelder konvertering fra MCI til demens, varierer tallene svært mye, og det er avgjørende om MCI (amnestisk MCI) er framkommet i en forskningspopulasjon (befolkningsundersøkelse) av eldre eller i en klinisk populasjon (pasienter ved en hukommelsesklinikk). I en populasjonsbasert studie (Ritchie, Artero & Tochon) ble det konkludert med at MCI var en dårlig prediktor for demens i løpet av en treårsperiode, med en 11 % konverteringsrate. Individene med MCI var også en ustabil gruppe, hvor nesten alle skiftet diagnostisk kategori hvert år. Det er også andre begreper (Kral, 1962; Crook et al., 1986; Levy, 1994) som er benyttet om kognitiv svikt hos eldre hvor det ikke er demens. For eksempel age-associated cognitive decline (AACD) (Levy, 1994), som heller er relatert til normal kognitiv aldring enn en påbegynnende demens. Diskriminant analyse viste ingen homogen klinisk gruppe. Imidlertid viste det seg at individer med AACD var langt mer stabile og i tillegg hadde en konverteringsrate til demens på nærmere 29 %. Dette var stikk i strid med hva en trodde på forhånd. I en oppsummeringsartikkel om MCI konkluderer DeCarli (2003) med at jo mer kognitiv svikt, bildediagnostikk og genetiske faktorer som trekker i retning av Alzheimers sykdom, desto mer sannsynlig er en snarlig progresjon fra MCI til demens ved Alzheimers sykdom. I forslaget til nye forskningskriterier for Alzheimers sykdom (DuBois et al., 2007) er dette konkretisert ved at man krever en kombinasjon av hukommelsessvikt og funn på biomarkører (MR, PET eller spinalvæskemarkører) for å stille diagnose.
Det er forsøkt behandling av MCI med kolinesterasehemmere (ChEls), uten at det kan sies å ha vært noen suksess. Raschetti, Albanese, Vanacore og Maggini (2007) oppsummerer i en oversiktsartikkel at ChEls ikke var assosiert med noen utsettelse på start av Alzheimers sykdom eller demens. Videre hadde en ikke full oversikt over sikkerhetsprofilen, det vil si mulige ulemper ved bruk av disse medikamentene. Det ble også satt spørsmålstegn ved den vitenskapelige validiteten ved studiene de gikk igjennom.
Normal aldring eller patologi?
I tittelen spør vi om disse kan skilles, men spørsmålet har liten mening for personer med en utviklet demens etter dagens kriterier som beskrevet ovenfor, og som åpenbart atskiller seg fra normale eldre. Det mer interessante spørsmålet er om en person med begynnende demensutvikling kan skilles fra en person med normale aldersforandringer, og om kognitive eller nevropsykologiske mål kan bidra til å skille.
Buckner (2004) kontrasterer de endringene i hjernen som er karakteristiske for normal aldring, og de som er typiske for Alzheimers sykdom, og foreslår at reduksjon i oppmerksomhet og eksekutiv funksjon er sentrale i normal aldring, mens reduksjon i hukommelse er karakteristisk for Alzheimers sykdom. Selv om det er en nyttig distinksjon, så er det et problem at det finnes flere typer av demens, spesielt noen former for vaskulær demens, hvor nettopp oppmerksomhet og eksekutiv funksjon er rammet tidlig i prosessen.
Ovenfor har vi diskutert MCI som en risikogruppe for demensutvikling. Dette er best dokumentert for den typen av MCI der hukommelse er sterkt involvert (amnestisk MCI), som synes å ha en klart øket risiko for progresjon til Alzheimers sykdom. Det er likevel slik at nevropsykologiske avvik alene ikke gir så sikker informasjon om begynnende demens at man kan stille en diagnose på nevropsykologisk grunnlag. Et problem med forskning på tidlige stadier av Alzheimers sykdom er også at diagnosekriteriene i dag krever at pasienten er dement, og at en sikker diagnose krever verifikasjon med obduksjonsmateriale. Samtidig er det meget sterke grunner til å tro at Alzheimers sykdom er en patologisk prosess som utvikler seg gjennom mange år før pasienten får en klinisk demensdiagnose. For å komme ut av dette uføret har en ekspertgruppe (DuBois et al., 2007) foreslått nye forskningskriterier for Alzheimers sykdom, der det ikke kreves at pasienten skal være dement for at man kan stille diagnosen. Man krever her redusert hukommelse som må dokumenteres med nevropsykologisk testing, men i tillegg funn på enten MR, PET eller spinalvæskeundersøkelse. Det er spesielt interessant at det nå er utviklet metoder for å påvise amyloide plakk i sentralnervesystemet, enten gjennom analyse av spinalvæske, som beskrevet ovenfor, eller ved å registrere en nyutviklet markør for PET-undersøkelser, der denne markøren (PIB) binder seg til amyloid og dermed tillater visualisering av amyloid opphopning (Klunk et al., 2004).
Nevropsykologisk påvisning av svikt er derfor fortsatt sentralt for å skille normal aldring fra tidlige stadier av patologi, spesielt ved Alzheimers sykdom. For å påvise svikt benytter vi oss av mål på avvik fra en norm, basert på data fra en referansegruppe. I det typiske referansematerialet som anvendes i dag, er det en klar økning i spredning av skårer med økende alder over ca. 60 år. Samtidig er referansematerialene basert på personer som anser seg selv som friske, men som ikke er grundig utredet med f.eks. MR av hjernen. Når vi vet at demenssykdommer utvikler seg langsomt over flere år, så er det svært sannsynlig at de referansegruppene som i dag danner grunnlag for våre normer for personer over 70 år, har et klart innslag av personer med tidlige stadier av patologi. Ved å anvende typiske grenseverdier for å vurdere avvik i form av 1,5 eller 2 standardavvik eller prosentiler (1. eller 5. prosentil) kan vi være temmelig sikre på at vi unngår å oppdage mange tilfeller av lettere patologi (Sliwinsky et al., 2003). Løsninger som er foreslått, er å bruke andre kutt-verdier (f.eks. 1 standardavvik) når man skal vurdere prestasjoner hos eldre, og å justere kutt-verdiene i forhold til den økte demensrisikoen som henger sammen med økende alder. En bedre løsning er muligens å bygge opp referansematerialer av personer som er grundig undersøkt, og der det også finnes longitudinelle data som garanterer at referansepersonene ikke utvikler demens i løpet av en rimelig oppfølgingstid av noen år. Det har også sine problemer, bl.a. at standarden for hva som er en grundig undersøkelse, er knyttet til teknologisk utvikling og derfor endrer seg raskt.
Avslutningsvis mener vi at nevropsykologisk undersøkelse bidrar til å identifisere personer med økt risiko for demensutvikling, og at svikt i hukommelse relativt til andre funksjoner gir grunn til mistanke. Ved vurdering av svikt bør vi ta et kritisk blikk på kvaliteten av det normative materialet som utgjør referansematerialet, og det er nødvendig med nevrologiske spesialundersøkelser dersom man skal si noe nærmere om årsak til kognitiv svikt.
Referanser
Anderson, L.A. & McConnell, S.R. (2007) The healthy brain and our aging population: Translating science to public health practise. Alzheimer's and Dementia, 3, 1 - 2.
Andersson, C. (2007). Predictors of cognitive decline in memory clinic patients. Doctoral thesis. Department of Neurobiology, Caring Sciences and Society Section of Clinical Geriatrics, Karolinska Institutet, Stockholm, Sweden.
Andersson, C., Lindau, M., Almkvist, O., Engfeldt, P., Johansson, S.-E., Eriksdotter Jönhagen, M. (2006). Identifying Patients at High and Low Risk of Cognitive Decline Using Rey Auditory Verbal Learning Test among Middle-Aged Memory Clinic Outpatients. Dement Geriatr Cogn Disorder, 21, 251 - 259.
Bartzokis, G. (2004) Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer's disease. Neurobiol Aging, 25, 5 - 18.
Blennow, K. & Vanmechelen, E. (2003). CSF markers for pathogenic processes in Alzheimer's disease: diagnostic implications and use in clinical neurochemistry. Brain Res Bull, 61, 235 - 242.
Braak, H. & Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl), 82, 239 - 259.
Buckner, R. (2004) Memory and executive function in aging and AD: Multiple factors that cause decline and reserve factors that compensate. Neuron, 44,195 - 208.
Cabeza, R., Anderson, N.D., Kester, J. & McIntosh A.R. (2002) Aging gracefully: compensatory brain activity in high-performing older adults. Neuroimage 17, 1394 - 1402.
Choi, D.W. (1992). Excitotoxic cell death. J Neurobiol, 23, 1261 - 1276.
Chua, H.F., Chen, W. & Park D.C. (2006). Source memory, aging and culture. Gerontology. 52, 306 - 313.
Collingridge, G,L. & Bliss, T.V. (1995). Memories of NMDA receptors and LTP. Trends Neurosci, 18(2), 54 - 56.
Cooper, R.J., Todd, J., McGill, K. & Michie P.T. (2006). Auditory sensory memory and the aging brain: A mismatch negativity study. Neurobiol Aging, 27,752 - 762.
Craik, F.I. & Bialystok, E. (2006). Cognition through the lifespan: mechanisms of change. Trends Cogn Sci.,10, 131 - 138.
Crook, T.H. & Ferris, SH. (1992). Age associated memory impairment.BMJ., 304, 714.
DeCarli, C. (2003). Mild cognitive impairment: prevalence, prognosis, aetiology, and treatment. Lancet Neurol, 2, 15 - 21.
de la Torre, J.C. (2002). Alzheimer's disease: how does it start?J Alzheimers Dis, 4, 497 - 512.
de la Torre, J.C. (2002). Vascular basis of Alzheimer's pathogenesis.Ann N Y Acad Sci, 977, 196 - 215.
de la Torre J.C. (2002). Alzheimer disease as a vascular disorder: nosological evidence.Stroke, 33, 1152 - 1162.
Dubois, B., Feldman, H., Jacova, C., DeKosky, S., Barberger-Gateau, P., Cummings, J., Delacourte, A., Galasko, D., Gauthier, S. & Jicha, G. (2007). Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurology, 6, 734 - 746.
Erkinjuntti, T. (2007). Vascular cognitive deterioration and stroke. Cerebrovascular Diseases, 24, 189 - 194.
Erkinjuntti, T., Lee, D.H., Gao, F., Steenhuis, R., Eliasziw, M., Fry, R., Merskey, H. & Hachinski, V.C. (1993). Temporal lobe atrophy on magnetic resonance imaging in the diagnosis of early Alzheimer's disease. Arch Neurol, 50, 305 - 310.
Erkinjuntti, T. & Hachinski, V.C. (1993). Rethinking vascular dementia. Cerebrovasc Dis, 3, 3 - 23.
Fjell, A.M., Walhovd, K.B., Reinvang, I., Lundervold, A., Dale, A. M., Quinn, B. T., Makris, N. & Fischl, B. (2005). Age does not increase rate of forgetting over weeks - Neuroanatomical volumes and visual memory across the adult life-span. Journal of the International Neuropsychological Society, 11, 1 - 14.
Fratiglioni, L., Launer, L.J., Andersen, K., Breteler, M.M., Copeland, J.R., Dartigues, J.F., Lobo, A., Martinez-Lage, J., Soininen, H. & Hofman, A. (2000). Incidence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurological Diseases in the Elderly Research Group. Neurology, 54, 10 - 15.
Furey-Kurkjian M.L., Pietrini, P., Graff-Radford, N., Alexander G.E., Freo, U., Szczepanik J. & Schapiro, M,B. (1995). Characterization of neuropsychological function in Alzheimer's disease and patients with prominent visual impairments. I: K. Iqbal., J.A. Mortimer, B. Winblad & H.M. Wisniewski (red.). Research Advances in Alzheimer's disease and related disorders. Chichester: Wiley, 1995: s. 225 - 234.
Gaeta, H., Friedman, D., Ritter, W. & Cheng, J. (1998). An event-related potential study of age-related changes in sensitivity to stimulus deviance. Neurobiol Aging, 19, 447 - 459
Galasko, D., Chang, L., Motter, R., Clark, C.M., Kaye, J., Knopman, D., Thomas, R., Kholodenko, D., Schenk, D., Lieberburg, I., Miller, B., Green, R., Basherad, R., Kertiles, L., Boss, M.A. & Seubert, P. (1998). High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol, 55, 937 - 945.
Graham, J.E, Rockwood, K., Beattie, B.L., Eastwood, R., Gauthier, S., Tuokko, H. & McDowell, I. (1997). Prevalence and severity of cognitive impairment with and without dementia in an elderly population. Lancet, 349, 1793 - 1796.
Guo, Z., Fratiglioni, L., Viitanen, M., et al. (2001). Apolipoprotein E genotypes and the incidence of Alzheimer's disease among persons aged 75 years and older: Variation by use of antihypertensive medication? Am J Epidemiol, 153, 225 - 231.
Gustafson L., Hagberg B. (1975). Dementia with onset in presenile period: a cross-sectional study. Acta Psychiatrica Scandinavica, 257, 38 - 71.
Hardy, J., Crook, R., Prihar, G., Roberts, G., Raghavan, R. & Perry, R. (1994). Senile dementia of the Lewy body type has an apolipoprotein E epsilon 4 allele frequency intermediate between controls and Alzheimer's disease. Neurosci Lett, 21,182, 1 - 2.
Head, D., et al. (2004) Differential vulnerability of anterior white matter in nondemented aging with minimal acceleration in dementia of the Alzheimer type: Evidence from diffusion tensor imaging. Cerebral Cortex, 14, 410 - 423.
Hestad, K. (1998). Den aldrende hjernen i nevropsykologisk perspektiv. Tidsskrift for Norsk Psykologforening, 35, 756 - 767.
Hestad, K., Dybing, E. & Kløve, H. (1997). Hukommelsestesting av eldre hvor det er mistanke om demens. Tidsskrift for Norsk Psykologforening, 35, 483 - 487.
Hestad, K. & Engedal, K. (2006). Antihypertensive Medication is Associated with Less Cognitive Impairment in the Very Old with Apolipoprotein-E e4 Allele. Drugs & Aging, 23, 723 - 731.
Hestad, K., Kveberg, B. & Engedal, K. (2005). Low blood pressure is a better predictor of cognitive deficits than the apolipoprotein e4 allele in the oldest old. Acta Neurologica Scandinavica, 111, 323 - 328.
Hoffman, A., Rocca, W.A., Brayne, C., et al., (1991). The prevalence of dementia in Europe: a collaborative study of 1980 - 1990 findings. International journal of epidemiology, 20, 736 - 748.
Kilander, L., Nyman, H., Boberg, M., Hansson, L. & Lithell, H. (1998). Hypertension is related to cognitive impairment. A 20-year follow-up of 999 men. Hypertension, 31, 780 - 786.
Kivipelto, M., Helkala, E-L., Hänninen, T., Laakso, M.P., Hallikainen, M., Alhainen, K., et al. (2001). Midlife vascular risk factors and late life cognitive impairment. A population-based study. Neurology, 56, 1683 - 1689.
Kivipelto, M., Helkala, E.L., Laakso, M.P., Hanninen, T., Hallikainen, M., Alhainen, K., Soininen, H., Tuomilehto, J. & Nissinen A. (2001). Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ, 322,1447 - 1451.
Kivipelto, M., Ngandu, T., Fratiglioni, L., Viitanen, M., Kareholt, I., Winblad, B., Helkala, E.L., Tuomilehto, J., Soininen, H. & Nissinen A. (2005). Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol, 62,1556 - 1560.
Korf, E.S., Wahlund, L.O., Visser, P.J. & Scheltens P. (2005). Medial temporal lobe atrophy on MRI predicts dementia in patients with mild cognitive impairment. Neurology, 63, 94 - 100.
Koutstaal, W. & Schacter, D.L. (1997) Gist-based false recognition for pictures in older and younger adults. Journal of Memory and Language, 37, 555 - 583.
Kral, V.A. (1962). Senescent forgetfulness: benign and malignant. Can Med Assoc J, 10, 257 - 260.
Kramer, A.F. & Kray. J. (2006). Aging and attention. I: F.I.M.Craik & E. Bialystok (Eds). Lifespan cognition. Oxford. Oxford University Press, s. 57 - 69.
Klunk, W.E., et al. (2004) Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol, 55, 306 - 319.
Launer, L.J., Masaki, K., Petrovik, H., Foley, D. & Havlik, R.J. (1995). The association between midlife BP levels and late-life cognitive function. The Honolulu-Asia Aging Study. JAMA, 274, 1846 - 1851.
Launer, L.J., Ross, G.W., Petrovitch, H., Masaki, K., Foley, D., White, L.R. & Havlik, R.J. (2000). Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol Aging, 21, 49 - 55.
Levy, R. (1994). Aging-associated cognitive decline. Working Party of the International Psychogeriatric Association in collaboration with the World Health Organization. Int Psychogeriatr, 6, 63 - 68.
Levy, R. Aging-associated cognitive decline. (1994). Erratum Int Psychogeriatr 1994, 6, 133.
Lewis, H., Beher, D., Cookson, N., Oakley, A., Piggott, M., Morris, C.M., Jaros, E., Perry R., Ince, P., Kenny, R.A., Ballard, C,G., Shearman, M.S. & Kalaria RN. (2006). Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol Appl Neurobiol, 32, 103 - 118.
Leys, D., Henon, H., Mackowiak-Cordoliani, M-A., Pasquier, F. (2005). Poststroke dementia. Lancet Neurol, 4, 752 - 759.
Lobo, A., Launer, L.J., Fratiglioni, L. et al. (2000). Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurology, 54, 4 - 9.
Mattson, M. & Magnus, T. (2006). Ageing and neuronal vulnerability. Nature Reviews Neuroscience, 7, 278 - 294.
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D. & Stadlan, E.M. (1984). Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology, 34, 939 - 944.
Nilsson, L.-G., et al. (1997). The Betula prospective cohort study: Memory, health and aging. Aging, Neuropsychology and Cognition, 1,1 - 32.
Murray, M.E,. Knopman, D.S., Dickson, D.W. (2007). Panminerva Med, 49, 197 - 207.
Oppenheim G. (1994). The earliest signs of Alzheimer's disease. J Geriatr Psychiatr Neurol, 7, 116 - 20.
O'Brien J.T. et al. (2003) Vascular cognitive impairment. Lancet Neurology, 2, 89 - 98.
Parasuraman, R. & Greenwood, P.M. (1998). Selective attention in aging and dementia. I: R. Parasuraman (Ed.) The attentive brain. Cambridge Mass, MIT press. s. 461 - 487.
Park, D.C. & Payer, D. (2006). Working memory across the adult lifespan. I: F.I.M.Craik & E. Bialystok (Eds). Lifespan cognition. Oxford. Oxford University Press, s. 128 - 139.
Park, D.C., Lautenschlager, G., Hedden, T., Davidson, N.S., Smith, A.D. & Smith, P.K. (2002) Models of visuospatial and verbal memory across the adult life span. Psychol Aging, 17, 299 - 320.
Parsons, C.G., Stoffler, A. & Danysz, W. (2007). Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system - too little activation is bad, too much is even worse. Neuropharmacology, in press.
Paxton, J.L., Barch, D.M., Racine, C.A. & Braver, T.S. (2007). Cognitive Control, Goal Maintenance, and Prefrontal Function in Healthy Aging. Cereb Cortex, Sep 17; [Epub ahead of print].
Peters, A., Morrison, J.H., Rosene, D.L. & Hyman, B.T. (1998). Are neurons lost from the primate cerebral cortex during normal aging? Cerebral Cortex, 8, 295 - 300.
Peters, A. & Sethares, C. (2002). Aging and the myelinated fibers in prefrontal cortex and corpus callosum of the monkey. J Comp Neurol 442, 277 - 291.
Peters, A. & Sethares, C. (2003). Is there remyelination during aging of the primate central nervous system? J Comp Neurol, 46, 238 - 254.
Petersen, R.C. (2004). Mild cognitive impairment as a diagnostic entity. J Internal Medicine, 256, 183 - 194.
Petersen, R.C., Smith, G.E., Waring, S.C., Ivnik, R.J., Kokmen, E. & Tangelos, E.G. (1997). Aging, memory, and mild cognitive impairment. Int Psychogeriatr, 9, 65 - 69.
Raschetti, R., Albanese, E., Vanacore, N. & Maggini, M. (2007). Cholinesterase Inhibitors in Mild Cognitive Impairment: A Systematic Review of Randomised Trials. PLoS Med, 4, 1818 - 1828.
Reinvang, I. (1999). Cognitive event-related potentials in neuropsychological assessment. Neuropsychology Review, 9, 231 - 248.
Riley, K.P., Snowdon, D.A., Desrosiers, M.F. & Markesbery, W.R. (2002). Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann Neurol, 51, 567 - 577.
Ritchie, K., Artero, S. & Touchon, J. (2001). Classification criteria for mild cognitive impairment: a population-based validation study. Neurology, 56, 37 - 42.
Roman, G.C., Tatemichi, T.K., Erkinjuntti, T., Cummings, J.L., Masdeu, J.C., Garcia, J.H., Amaducci, L., Orgogozo, J.M., Brun, A., Hofman, A., et al. (1993). Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology, 43, 250 - 260.
Rogers, S.L., Farlow, M.R., Doody, R.S., Mohs, R. & Friedhoff, L.T. (1998) A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer's disease. Donepezil Study Group. Neurology, 50,136 - 45. Comment in: Neurology, 1999 1, 52, 218 - 219.
Salthouse, T.A. (1996). The processing-speed theory of adult age differences in cognition. Psychol Rev. 103, 403 - 428.
Skoog, I. & Gustafson, D. (2003). Hypertension, hypertension-clustering factors and Alzheimer's disease. Neurol Res, 25, 675 - 680.
Sliwinsky, M., Lipton, R., Buschke, H. & Wasylyshyn, C. (2003). Optimizing cognitive test norms for detection. I: R.C.Petersen (Ed) Mild cognitive impairment.. Oxford, Oxford University Press, s. 89 - 104.
Spencer, W.D. & Raz, N. (1995). Differential effects of aging on memory for content and context: A meta-analysis. Psychology and Aging, 10, 527 - 539.
Strittmatter, W.J., Saunders, A.M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G.S. & Roses, A.D. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A, 1, 90, 1977 - 1981.
Suzuki, K., Kutsuzawa, T., Nakajinia, K. & Hatano, S. (1991). Epidemiology of vascular dementia and stroke in Japan. I: Hartmann A, Kuschinsky W, Hoyer S. (red.). Cerebral Ischemia and Dementia. Berlin: Springer-Verlag; 1991. s. 16 - 24.
Swan, G.E., DeCarli, C., Miller, B.L., Reed, T., Wolf, P.A, Jack, L.M. & Carmelli, D. (1998). Association of midlife blood pressure to late-life cognitive decline and brain morphology. Neurology, 51, 986 - 993.
Swan, G.E, Carmelli, D. & Laurue, A. (1998). Systolic blood pressure tracking over 25 to 30 years and cognitive performance in older adults. Stroke, 29, 2334 - 2340.
Tatemichi, T.K., Desmond, D.W., Stern, Y., Sano, M., Mayeux, R., Andrews, H. (1992). Prevalence of dementia after stroke depends on diagnostic criteria. Neurology, 42, 413.
Tatemichi, T.K., Desmond, D.W., Mayeux, R., et al. (1992). Dementia after stroke: baseline frequency, risks, and clinical features in a hospitalized cohort. Neurology, 42, 1185 - 1193.
Tranel, D. (2002). Emotion, decision making, and the ventromedial prefrontal cortex. I: D.T.Stuss & R.T.Knight. (Ed) Principles of frontal lobe function. Oxford, Oxford University Press, s. 338 - 353.
Walhovd, K. B., Fjell, A.M., Reinvang, I., Lundervold, A., Dale, A. M., Eilertsen D.E., Quinn, B. T. Salat, D. & Fischl, B. (2005). Effects of age on volumes of cortex, white matter and subcortical structures. Neurobiology of Aging, 26, 1261 - 1270.
West, R.L. (1996). An application of prefrontal cortex function theory in cognitive aging. Psychological Bulletin, 120, 272 - 292.
WHO. The ICD-10 classification of mental and behavioural disorders: diagnostic criteria for research. Geneva: WHO, 1993.
Wu, C.C., Mungas, D., Petkov, C.I., Eberling, J.L., Zrelak, P.A., Buonocore, M.H., Brunberg, J.A., Haan, M.N. & Jagust, W.J.. (2002). Brain structure and cognition in a community sample of elderly Latinos. Neurology, 59, 383 - 391.Vitenskap og psykologi