")
Du er her
Epigenetiske endringer ved alvorlige psykiske lidelser - et resultat av psykososialt stress?
Abstract:
Epigenetic changes in severe mental illness: a result of psychosocial stress?A challenge for environmentally oriented theories of mental illness etiology has been to delineate the mechanisms by which exposure to psychosocial stress changes neurobiological structure and function. One hypothesis is that epigenetic mechanisms play a role. The scientific literature was reviewed in order to assess the hypothesis that epigenetic changes induced by psychosocial stress are involved in severe mental illness (schizophrenia, bipolar disorder and major depression), with a focus on studies of DNA methylation in the brain using post-mortem samples. An array of DNA methylation changes has been reported in severe mental illness, but with large variability across studies. Most importantly, candidate gene studies of epigenetic alterations in severe mental illness generally have not focused on genes that are central to stress processing, and epigenome-wide association studies (EWAS) have not targeted epigenomes derived from brain regions that are central to stress-response systems. The available evidence currently does not allow for the evaluation of whether stress-induced epigenetic changes characterize severe mental illness. |
Keywords: Epigenetics, DNA methylation, psychosocial stress, severe mental illness, developmental trauma |
Psykososiale påvirkninger som oppleves å true ens velvære, helse eller liv (psykososialt stress) aktiverer en rekke systemer i hjernen og kroppen (Koolhaas et al., 2011). Sentralt er den nevroendokrine hypothalamus-hypofyse-binyre (HPA)-aksen som skiller ut stresshormoner som kortiotrofinutløsende hormon (CRH), adrenokortikotrofisk hormon (ACH) og kortisol; det autonome nervesystemet som skiller ut adrenalin og noradrenalin; samt dopaminsystemet og immunsystemet. Påvirkninger som kun er moderat stressende og knyttet til mestringsfølelse, kan være positive for ens utvikling (eustress) (Selye, 1974). Nevrobiologisk kjennetegnes slik mestring ved at reguleringsområder i forhjernen som hippocampus og prefrontal cortex spiller tilbake på og bremser aktiviteten i stressresponssystemene. Psykososialt stress kan imidlertid bli toksisk når reguleringsområdene ikke får bremset aktiviteten i stressresponssystemene, slik som ved gjentatt eksponering for alvorlige seksuelle og fysiske overgrep, emosjonelle overgrep, emosjonell neglisjering og mobbing i oppveksten. Dette er en av mekanismene bak det Nordanger og Braarud (2017) refererer til som et svekket «reguleringssystem» hos barn og unge som er utsatt for utviklingstraumer. Når aktiveringen av stressresponssystemene vedvarer over tid i oppveksten, ses gjerne både funksjonelle og strukturelle endringer i hjernen. HPA-stressaksen kan bli mer sensitiv for nye, potensielt truende erfaringer, men samtidig mindre reaktiv på kjente stressorer (Herman et al., 2016). Hippocampus og midtre deler av prefrontal cortex kan krympe og bli mindre, mens amygdala typisk vokser i størrelse for senere å krympe (Caspi et al., 2003; Herman et al., 2016; Pruessner, Cullen, Aas, & Walker, 2017; Read, Fosse, Moskowitz, & Perry, 2014; Teicher & Samson, 2016; Walker & Diforio, 1997). I tillegg har eksperimentell forskning på dyr avdekket en rekke detaljerte endringer etter toksisk psykososialt stress tidlig i livet. Dette inkluderer tap av dendritter (fangarmer) på nervecellene og endrede synapseforbindelser i hippocampus og prefrontal cortex, vekst i dendrittene i amygdala og hemmet nevrogenese i hippocampus (McEwen et al., 2015).
Endringene som typisk ses i hjernen etter toksisk psykososialt stress eller utviklingstraumer, sammenfaller med de som ses ved alvorlige psykiske lidelser, som (alvorlig) depresjon, bipolar lidelse og schizofreni. Dette inkluderer endret størrelse på prefrontal cortex, hippocampus og amygdala, en mer sensitiv og/eller mindre reaktiv HPA-akse og detaljerte endringer i nervecellene, som vekst eller reduksjon av dendrittrær (Read et al., 2014; Teicher & Samson, 2016). Alvorlige psykiske lidelser er også assosiert med høye forekomster av alvorlig psykososialt stress i oppveksten. Samlet støtter disse funnene hypotesen om at psykososialt stress ligger til grunn for endringene i hjernen ved disse lidelsene (Read et al., 2014; Teicher & Samson, 2016). En utfordring med denne hypotesen er imidlertid at det er uklart hvilke molekylære mekanismer som ligger til grunn for nevrobiologiske endringer etter utviklingstraumer. De siste årene har epigenetiske mekanismer blitt lansert som en mulig forklaring, ettersom slike mekanismer både påvirkes av våre erfaringer og kan modifisere genenes aktivitetsnivå (McGowan & Szyf, 2010; Nestler, Pena, Kundakovic, Mitchell, & Akbarian, 2015). I denne artikkelen vil jeg vurdere om forskningen støtter en hypotese om at epigenetiske mekanismer er endret på samme måte ved alvorlige psykiske lidelser som etter alvorlig psykososialt stress tidlig i livet.
Epigenetisk regulering av genene
På et grunnleggende nivå reguleres strukturen og funksjonen til hjernen og kroppens organer (fenotypene) av genuttrykket. Det første skrittet i genuttrykket er transkripsjon av RNA (ribonukleinsyre), der et gen (en sekvens av basepar på DNA) kopieres til RNA. Deretter modifiseres RNA til budbringer-RNA, som så transporteres ut av cellekjernen. Vel ute av cellekjernen oversettes budbringer-RNA til aminosyrekjeder, som i siste instans folder seg til proteiner. Proteinene er våre biologiske byggeklosser, slik at hjernens struktur og funksjon hviler på den relative produksjonen av ulike proteiner i de 80–90 milliarder nervecellene i hjernen.
Dyreforskningen har dokumentert at alvorlig psykososialt stress påvirker genuttrykket i en rekke systemer og områder i hjernen (Deak et al., 2015; Tung & Gilad, 2013). For eksempel er alvorlig psykososialt stress knyttet til redusert uttrykk av genet for glukokortikoidreseptoren (GR) for kortisol (NR3C1-genet), slik at det blir færre slike reseptorer i hippocampus og prefrontal cortex (Chiba et al., 2012; Patel, Katz, Karssen, & Lyons, 2008). Andre gener som uttrykker seg annerledes etter psykososialt stress, inkluderer gener for hjerneavledet nevrotropisk faktor (brain derived neurotrophic factor, BDNF, involvert i nevral vekst), proinflammatorisk aktivitet i immunsystemet, GABA-internevronfunksjon og glutamatfunksjon (Tung & Gilad, 2013; Uchida, Furukawa, Iwata, Yanagawa, & Fukuda, 2014; Veeraiah et al., 2014). I hippocampus alene ses endret uttrykk av flere hundre gener etter alvorlig psykososialt stress, der grad og varighet avhenger av karakteristika ved stressoren, individets læringshistorie, følelsen av mestring og den psykososiale konteksten (McEwen et al., 2015). Det endrete genuttrykket etter psykososialt stress omtales i biologien gjerne som sentralt for vår tilpasning (adaptasjon, fenotypisk plastisitet) til endringer i det ytre miljøet (Weaver, 2014).
Epigenetikk (prefikset epi er gresk for over, utenfor eller rundt) er kjemiske markører som omgir DNA i cellekjernen, og som påvirker genuttrykket. Etter at begrepet først ble introdusert på 1940-tallet (Waddington, 1942), har det blitt definert på ulike måter. I dag defineres epigenetikk gjerne som endringer i genuttrykket som ikke skyldes variasjon i selve DNA-sekvensen, men i stedet andre faktorer i kromatinet i cellekjernen (Deans & Maggert, 2015; Mitchell, Roussos, Peter, Tsankova, & Akbarian, 2014). I tillegg til DNA består kromatinet av kjemiske markører som kan binde seg til DNA, og av en ball-liknende molekylær struktur, såkalte nukleosomer, som DNA er kveilet rundt (Luger, Mader, Richmond, Sargent, & Richmond, 1997) (figur 1).
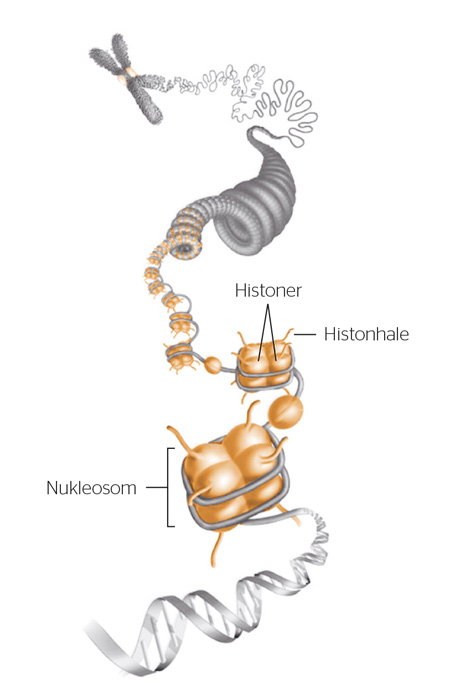
FIGUR 1. Kromatinets organisering i kromosomene
Den epigenetiske mekanismen som har vært klart mest studert ved psykiske lidelser, er CG-metylering. Ved CG-metylering binder en metylgruppe seg til karbon på cytosinbasen i et cytosin-guanin-basepar (CG) på DNA (figur 2). CG-basepar forekommer både spredt utover DNA og klumpet sammen i «øyer». Ca. 60 % av våre rundt 20 000 proteinproduserende gener har CG-rike øyer (Antequera & Bird, 1993; Venter et al., 2001). Slike øyer sammenfaller ofte med promoterområder til gener. Dette er startsteder for transkripsjonen av DNA til RNA, og disse områdene er sentrale i den epigenetiske forskningen (Saxonov, Berg, & Brutlag, 2006). Cytosinmetylering av CG-basepar på promoterområdet til et gen hindrer vanligvis genuttrykket, slik at hypermetylerte gener har et dempet uttrykk (Jones, 2012). CG-metylering hindrer genuttrykket både ved å blokkere tilgangen til maskineriet som transkriberer gener, og ved å tiltrekke seg proteiner som lukker kromatinet rundt et gen, det vil si får nukleosomene til å pakke seg tett sammen (Viana et al., 2016). Motsatt vil tap av metylering ofte føre til økt gentranskripsjon.
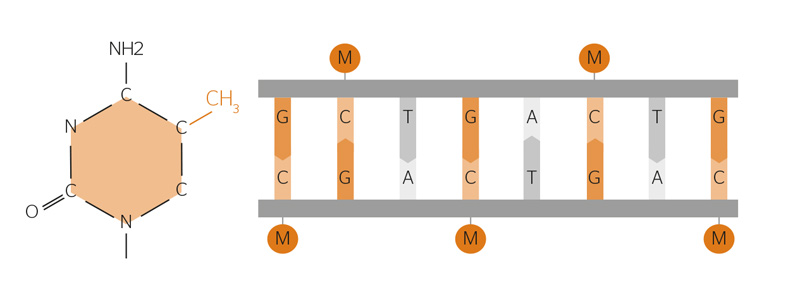
FIGUR 2. CG-metylering
Til høyre: En sekvens av DNA med baseparene cytosin-guanin (CG) og adenin-thymin (AT) samt metylering av cytosin (M). Til venstre: Cytosinmetylering ses når en metylgruppe (CH3) binder seg kovalent (elektronbinding) til et karbonatom (C) på cytosinmolekylet. Dette hindrer at transkripsjonsfaktorer binder seg til DNA, og dermed også genuttrykket. Figuren er nedlastet fra http://www.sigmaaldrich.com/technical-documents/articles/biofiles/introd…
Formen på kromatinet kan også modifiseres ved at kjemiske markører binder seg til «histonhaler», kjeder av aminosyrer som løper ut fra nukleosomene, «ballene» som DNA er kveilet rundt (figur 1). Kjemiske bindinger til histonhalene gjør kromatinet mer eller mindre kompakt, noe som modifiserer hvor tilgjengelig genene er for transkripsjon (Encode Project Consortium, 2007). Histonmodifiseringer har til nå blitt forholdsvis lite studert for psykiske lidelser.
Gjennom individets prenatale utvikling reguleres prosessen med celledifferensiering i stor grad av nedarvede epigenetiske mekanismer (Meissner, 2010). Mens alle cellene i kroppen og hjernen i hovedsak har samme DNA-sekvens, er det epigenetiske markører på flere tusen steder på genomet som bestemmer hvilke gener som er aktive i ulike celler, og dermed hva slags celle som utvikles, for eksempel en gitt type nervecelle eller en hudcelle. Prenatalt er de epigenetiske mekanismene høyst sensitive på det ekstracellulære miljøet, herunder mors ernæring, psykologiske tilstand og atferd slik dette påvirker fosteret via morkaken (Kundakovic & Jaric, 2017). Postnatalt og gjennom oppveksten er epigenetiske mekanismer sentrale både for normalutvikling, som forutsetter spesifikke miljøerfaringer i sensitive tidsvinduer, og for utvikling og tilpasning basert på stresseksponering (Bock, Rether, Groger, Xie, & Braun, 2014). Hjerneprosesser som utvikler seg i dette samspillet, inkluderer modning av nervecellenes dendritter og synaptiske forbindelser. Dette ligger igjen til grunn for utviklingen av funksjonalitet i og mellom komplekse nevrale nettverk, herunder hippocampus, amygdala og prefrontal cortex og for interaksjonen mellom disse områdene og HPA-stress-aksen.
Problemstilling
I denne artikkelen undersøkes om forskningen som er utført til nå, støtter hypotesen om at epigenetiske endringer ved alvorlige psykiske lidelser er stressbaserte. Forskning på epigenetiske endringer, særlig CG-metylering, etter psykososialt stress og ved schizofreni, bipolar lidelse og alvorlig depresjon oppsummeres og sammenliknes.
Metode
Mye av forskningen på epigenetiske endringer etter stress og ved psykiske lidelser har vært basert på prøver fra perifert vev, som blod og spytt (Cariaga-Martinez & Alelu-Paz, 2016). Epigenetiske markører varierer imidlertid betydelig mellom perifert vev og hjernen så vel som mellom ulike strukturer i hjernen (Bakulski, Halladay, Hu, Mill, & Fallin, 2016; Cariaga-Martinez & Alelu-Paz, 2016; Viana et al., 2016). Det er omdiskutert om epigenetiske tilstander i perifert vev kan gi informasjon om tilstander i hjernen generelt og i spesifikke hjernestrukturer spesielt (Jaffe & Kleinman, 2016; Walton et al., 2016). Av denne grunn avgrenses gjennomgangen til studier basert på prøver fra hjernen. Dette tilsier post mortem-studier av mennesker, i tillegg til eksperimentelle studier av dyr som har blitt utsatt for psykososialt stress i oppveksten og deretter ofret og dissekert.
Litteratursøk ble utført i PubMed, med inklusjon av artikler publisert frem til medio februar 2017. Følgende hovedsøkestreng ble brukt: epigen*[Title] OR methylation[Title] OR histone[Title]. Denne hovedsøkestrengen ble komplementert med følgende søkestrenger for de to undertemaene som dekkes i artikkelen: epigenetiske endringer etter psykososialt stress – AND (stress[Title] OR trauma*[Title] OR abuse[Title] OR neglect[Title] OR maltreatment[Title] OR «maternal care»[Title]); samt epigenetiske endringer ved alvorlige psykiske lidelser – AND (mental[Title] OR psychopathol*[Title] OR psychiatr*[Title] OR psychosis[Title] OR psychotic[Title] OR schizophren*[Title] OR depression[Title] OR depressive[Title] OR bipolar[Title]). Referanselistene i originalartikler og nyere oversiktsartikler ble også gjennomgått for å identifisere relevant litteratur.
I oppsummeringen av forskningen refereres kun statistisk signifikante funn. I studier som har undersøkt epigenetiske endringer på hele genomet i epigenomvide assosiasjonsstudier (EWAS), gjengis kun funn som nådde et genomvidt nivå for statistisk signifikans etter kontroll for multiple tester.
Resultater
Epigenetiske endringer etter psykososialt stress
Eksperimentell forskning på dyr indikerer at prenatalt stress via mor kan endre epigenetiske markører i en rekke områder i hjernen og modifisere avkommets nevrobiologiske utvikling (Griffiths & Hunter, 2014). Kandidatgenstudier (analyser av spesifikke gener) indikerer endret CG-metylering etter prenatalt stress av både NR3C1-genet for kortisolreseptoren GR og av et promoterområde til et gen for kortikotrofinutløsende hormon. For begge genene har forskerne funnet endret CG-metylering i både prefrontal cortex, hippocampus, amygdala og hypothalamus hos avkommet. Slike metyleringsendringer er også knyttet til endringer i både genuttrykket og i atferden til avkommet etter fødselen, så som økt stress-sensitivitet (Mueller & Bale, 2008; Mychasiuk, Ilnytskyy, Kovalchuk, Kolb, & Gibb, 2011). Dyrestudiene indikerer også endret metylering av andre gener hos avkommet etter prenatalt stress, som av promoterområder til et gen for reelin (knyttet til kortikal og synaptisk modning) og GAD67 (knyttet til GABA internevronfunksjon) i prefrontal cortex (Matrisciano et al., 2013; Palacios-Garcia et al., 2015). Ingen studier synes å foreligge om epigenetiske endringer i hjernen hos mennesker etter prenatalt psykososialt stress via mor.
Fra 1990-tallet og fremover har forskning på gnagere vist hvordan psykososialt stress i oppveksten kan forme hjerneutvikling og atferd ved å modifisere epigenetiske mekanismer (Weaver, 2009; Weaver et al., 2004). Forskerne har tatt utgangspunkt i at høy grad av slikking og stelling av avkommet (god morsomsorg) gjerne fører til trygg og modig atferd, mens lite slikking og stelling (neglisjering) fører til høy stress-sensitivitet, svekket læring og hukommelse og engstelig atferd. Kontrollerte kryssfostringseksperimenter viste at neglisjering førte til tap av kortisolreseptoren GR i hippocampus, og at dette hadde et epigenetisk grunnlag; promoterområder for NR3C1 GR-genet var mer metylert hos de neglisjerte rottene (Meaney & Szyf, 2005). Påfølgende studier av gnagere har indikert en rekke andre epigenetiske endringer etter relasjonsstress i oppveksten. Dette inkluderer CG-metylering og/eller histonmodifiseringer for gener involvert i reguleringen av CRH, vasopressin og østrogen i hypothalamus, GABA-reseptorer og glutamatreseptorer i hippocampus, nevrotensinreseptorer i amygdala, hjerneavledet nevrotropisk faktor (BDNF) i prefrontal cortex, cannabinoide reseptorer i cingulum cortex og serotoninreseptoren 5HT1A i prefrontal cortex og midthjernen (Jawahar, Murgatroyd, Harrison, & Baune, 2015; Kundakovic & Champagne, 2015; Le Francois et al., 2015; Lomazzo, Konig, Abassi, Jelinek, & Lutz, 2017; Provencal et al., 2012). I en eksperimentell studie på rhesusapen analyserte forskerne metylering av genpromoterområder på hele genomet i en EWAS. Tidlig relasjonsstress var her knyttet til endret metylering av en lang rekke gener i prefrontal cortex, inklusive gener involvert i stressresponser, responser på andre typer stimuli, immunsystemfunksjon, vekst i aksoner og utvikling av nervesystemet (Provencal et al., 2012). Et slikt spekter av metyleringsendringer kan representere en koordinert systemeffekt der hjernen som helhet tilpasses viktige sider ved individets livshistorie, som tidlig relasjonsstress (Zhang, Labonte, Wen, Turecki, & Meaney, 2013).
I litteraturutvalget var det kun én forskergruppe som hadde undersøkt sammenhengen mellom psykososialt stress og epigenetiske endringer i hjernen hos mennesker (Labonte et al., 2012; McGowan et al., 2009). I en genkandidatstudie undersøkte disse forskerne epigenetiske markører for NR3C1 GR-genet for kortisol i hippocampus hos personer som hadde begått selvmord og erfart alvorlig relasjonsstress i oppveksten (seksuelle overgrep, alvorlige fysiske overgrep eller alvorlig neglisjering). Forskerne rapporterte at de avdøde som var utsatt for alvorlig relasjonsstress i oppveksten, hadde økt CG-metylering av et promoterområde til NR3C1-genet for GR, sammenliknet med en matchet gruppe avdøde uten slike erfaringer. Som i dyreforskningen var den økte metyleringen knyttet til et lavere uttrykk av NR3C1-genet. Forskergruppen fulgte opp genkandidatstudien med en EWAS og fant at relasjonsstress tidlig i livet var knyttet til endret metylering av 307 promoterområder i hippocampus (Labonte et al., 2012). Endringene inkluderte både hyper- og hypometylering, herunder for gener involvert i nevral plastisitet og immunsystemfunksjon.
Epigenetiske endringer ved alvorlige psykiske lidelser
Ved alvorlige psykiske lidelser er epigenetiske endringer i post mortem-hjernevev, i hovedsak CG-metylering, undersøkt med både genkandidatstudier og EWAS. De fleste av studiene har fokusert på schizofreni og noen få på bipolar lidelse og alvorlig depresjon.
Genkandidatstudier
I genkandidatstudier er det rapportert om epigenetiske endringer på promoterområder for gener involvert i serotoninfunksjon i prefrontal cortex ved både schizofreni og bipolar lidelse. Mens metyleringsendringer etter psykososialt stress hos gnagere er rapportert for serotonin 1A-reseptoren, fant Abdolmaleky og kollegaer endret metylering på gener for 2A-reseptoren og serotonintransportøren 5-HTT ved schizofreni og bipolar lidelse (Abdolmaleky et al., 2014; Abdolmaleky et al., 2011). Videre rapporterte Tang, Dean og Thomas (2011) om histonmodifiseringer for serotonin 2C-reseptoren ved schizofreni. Enkelte studier har indikert metyleringsendringer på reelingenet i prøver fra prefrontal cortex ved schizofreni, noe som også er rapportert etter prenatalt stress hos dyr, men andre studier har ikke kunnet replikere dette (Abdolmaleky et al., 2005; Mill et al., 2008; Tochigi et al., 2008). Tilsvarende er det rapportert om endret metylering i prefrontal cortex for et gen som koder for BDNF ved schizofreni, noe som også er indikert i dyreforskningen på psykososialt stress, men heller ikke dette er et konsistent funn (Cheah et al., 2016; Keller et al., 2014; Weickert et al., 2003). En genkandidatstudie identifiserte endret metylering av MB-COMT i prefrontal cortex ved schizofreni, et gen som er knyttet til dopaminfunksjon (Abdolmaleky et al., 2006). Metylering av dopamingener ser ikke ut til å ha vært fokusert på i stressforskningen, selv om dopaminsystemet aktiveres og endres av alvorlig psykososialt stress (Holly & Miczek, 2016). Ingen post mortem genkandidatstudier ser ut til å ha fokusert på epigenetiske endringer i HPA-aksen, herunder NR3C1 GR-genet, ved alvorlige psykiske lidelser.
Epigenomvide assosiasjonsstudier
I den første EWAS for schizofreni og bipolar lidelse rapporterte Mill og medarbeidere (2008) om endret CG-metylering av over 50 gener i prefrontal cortex, herunder gener implisert i GABA- og glutamatfunksjon, noe som også er funnet etter psykososialt stress, samt gener knyttet til dopaminsystemet, immunsystemet, celleproliferering og differensiering, og utvikling av hjernen. I en annen EWAS fant Pidsley og medarbeidere (2014) endret metylering av fire områder på DNA i prefrontal cortex hos avdøde diagnostisert med schizofreni, herunder av HTR5A-genet, som koder for serotoninreseptoren 5A og for Neuritin 1-genet, som er knyttet til synaptisk plastisitet og utvikling av nervesystemet. Det var ingen overlapp mellom funnene i denne studien og funnene til Mill og medarbeidere (2008). Numata og medarbeidere (2014), som også studerte schizofreni, rapporterte om endret metylering av 107 CG-posisjoner i prøver hentet fra prefrontal cortex, herunder på et gen som koder for en glutamatreseptor, men der kun ett av områdene var indikert i tidligere studier. Dette var for et gen for mitokondriefunksjon som også ble rapportert av Mill og medarbeidere (2008). Wockner og medarbeidere (2014) identifiserte endret CG-metylering av hele 2929 gener i fremre hjernebark ved schizofreni. Kun ett av disse genene var indikert også i tidligere studier, FAM5C-genet knyttet til celleproliferering og migrasjon (Numata et al., 2014). Blant de mange signifikante funnene fremhevet Wockner og medarbeidere (2014) gener knyttet til gliacellefunksjon og 1A-reseptoren for serotonin. Også Jaffe og medarbeidere (2016) rapporterte om et stort antall områder på DNA (n = 2104) med endret CG-metylering ved schizofreni, med prøver tatt fra dorsolateral prefrontal cortex. Jaffe og medarbeidere (2016) var sparsomme med informasjon om hvilke gener det handlet om, men fremhevet gener knyttet til tidlig utvikling av fosteret, utvikling og spesialisering av celler og differensiering i nervesystemet. I en studie publisert i 2016 analyserte Viana og medarbeidere (2016) prøver hentet fra prefrontal cortex, hippocampus og striatum ved schizofreni. De fant 12 statistisk signifikante CG-posisjoner med endret metylering i de tre hjerneområdene. Blant disse var endret metylering av gener knyttet til samspillet mellom nerveceller og til synaptisk plastisitet i prefrontal cortex, proteinmetabolisme i hippocampus og formen og mobiliteten til nerveceller i striatum. Funnene i EWAS-forskningen til nå er dermed noe sprikende for schizofreni, mens lite har vært undersøkt når det gjelder bipolar lidelse.
Den første post mortem-EWAS for alvorlig depresjon ble publisert i 2012 (Sabunciyan et al., 2012). Ingen posisjoner på DNA, hentet fra prefrontal cortex, nådde her et epigenomvidt signifikansnivå. Forfatterne foreslo at videre forskning burde se på andre hjerneområder enn prefrontal cortex, som anterior cingulate, hippocampus og amygdala, samt være mer spesifikke på hvilke typer nerveceller som studeres. Haghighi og kolleger (2014) studerte metyleringsmønstre i orbitofrontal cortex (en del av prefrontal cortex) hos pasienter med alvorlig depresjon som hadde begått selvmord. Deres hovedkonklusjon var at CG-metylering i dette hjerneområdet først og fremst varierte med deltakernes alder. I tillegg fant de åtte ganger så mange metylerte CG-posisjoner i suicid-/depresjonsgruppen som i en kontrollgruppe, blant annet for gener knyttet til cellulær utvikling, overlevelse og død. Disse positive assosiasjonene ble ikke replisert i den siste (per februar 2017) EWAS for alvorlig depresjon, som igjen fokuserte på prefrontal cortex. Denne studien identifiserte ingen områder på DNA med signifikant endret CG-metylering (Murphy et al., 2017).
Diskusjon
Forskning særlig på gnagere har indikert at psykososialt stress tidlig i livet kan endre CG-metylering av gener i HPA-aksen, som NR3C1 for GR, og av en rekke gener i hypothalamus, hippocampus og prefrontal cortex. Post mortem-studier indikerer at også hos mennesker kan psykososialt stress føre til endret metylering av sentrale gener i HPA-aksen og gi omfattende metyleringsendringer på genomet i hippocampus. Endret CG-metylering av gener for nevrobiologiske komponenter som er omtalt i stressforskningen, har også blitt rapportert i enkelte studier av alvorlige psykiske lidelser, særlig schizofreni. Dette inkluderer gener for synaptisk plastisitet, nevrobiologisk utvikling, glutamat- og GABA-funksjon, serotoninfunksjon, BDNF, reelin og immunsystemet. Men de identifiserte genene er ofte ikke de samme som i stressforskningen, og funnene har sjelden blitt replikert i påfølgende studier.
Forskningen på epigenetikk ved alvorlige psykiske lidelser synes i liten grad å ha vært utført innenfor et psykososialt stressperspektiv. Ingen hypotesedrevne studier (genkandidatstudier) for alvorlige psykiske lidelser har fokusert på epigenetisk modifisering av de genene som er mest sentrale i hjernens stressresponssystemer. I påfølgende hypotesefrie studier der hele genomet har vært skannet i EWAS, har analysene ikke vært basert på vevsprøver fra hjerneområder som er eksplisitt involvert i prosesseringen av stress. Dette er et essensielt problem fordi epigenetiske markører generelt så vel som ved psykiske lidelser ser ut til å variere betydelig ikke bare mellom hjernen og perifere mål slik som blodet og huden, men også mellom ulike områder i hjernen (Davies et al., 2012; Kozlenkov et al., 2016). Ved schizofreni og bipolar lidelse er for eksempel epigenetiske markører rapportert å være endret på svært ulike måter i prefrontal cortex og anterior cingulate (Xiao et al., 2014). I forskningen på alvorlige psykiske lidelser som ble identifisert i litteratursøket, oppgav forfatterne stort sett kun at vevsprøver var hentet fra prefrontal cortex. Det er høyst uklart om denne forskningen gir informasjon om epigenetiske endringer i de mest stressrelevante hjerneområdene, for eksempel de spesifikke cellegruppene som regulerer HPA-stressaksen i hypothalamus, hippocampus, infralimbiske deler av den fremre hjernebarken og amygdala (Herman et al., 2016). Den gjennomgåtte forskningen er snarere utført innenfor et biogenetisk perspektiv enn et stressperspektiv, der vekten har vært på å undersøke om det er strukturelle variasjoner på DNA (heller enn stress) som er årsaken til metyleringsendringer ved alvorlige psykiske lidelser (se f.eks. Jaffe et al., 2016; Viana et al., 2016). Hvis en antar at strukturell variasjon på DNA er det som styrer epigenetiske endringer ved psykiske lidelser, kan det vurderes som mindre vesentlig nøyaktig hvor vevsprøver hentes fra. Men for at forskningen skal bli relevant i et stressperspektiv, bør den baseres på vevsprøver fra hjerneområder som er involvert i prosesseringen av stress. Selv dette kan vise seg å være en for grovkornet innfallsvinkel, og epigenom bør kanskje hentes fra spesifikke typer celler (f.eks. pyramideceller, en gitt type internevroner) i underområder i hjernen som er høyst presist avgrenset med tanke på funksjon. Teknologiske nyvinninger indikerer at et slikt presisjonsnivå i forskningen nå kan være mulig på post mortem-hjerner (Heijmans & Mill, 2012; Jaffe, 2016).
De fleste studiene av epigenetiske endringer etter psykososialt stress og ved alvorlige psykiske lidelser har fokusert på CG-metylering. En utfordring er at selv om CG-metylering anses som den mest stabile epigenetiske markøren, er den likevel forholdsvis dynamisk og endrer seg markant fra oppveksten til voksent liv og alderdommen (Haghighi et al., 2014). Dermed kan det være vanskelig å identifisere tidlige metyleringsendringer etter psykososialt stress flere tiår senere i post mortem-studier (Heijmans & Mill, 2012). Andre kompliserende forhold er at epigenetiske markører påvirkes av faktorer som medisinering og røyking (som ses oftere ved alvorlige psykiske lidelser) og av tidsintervallet fra død inntreffer til nedfrysning av hjernen og prøver blir analysert (Monoranu et al., 2011). I tillegg fordrer en helhetlig forståelse av epigenetiske endringer ved både stress og alvorlige psykiske lidelser at det også forskes på andre mekanismer enn CG-metylering. Flere andre mekanismer bidrar til å regulere genuttrykket, som en rekke former for DNA-metylering utover CG-metylering, histonmodifiseringer, visse typer RNA og en lang rekke andre kjemiske markører og mekanismer som påvirker kromatinets tilstand. Forståelsen av alle disse komponentene og av samspillet mellom dem utgjør et viktig fokus i kommende forskning (Goldberg, Allis, & Bernstein, 2007; Mitchell et al., 2014; Roadmap Epigenomics et al., 2015).
Konklusjon
Epigenetiske endringer ved alvorlige psykiske lidelser er et nytt forskningsområde som foreløpig ikke har vært orientert mot et psykososialt stressperspektiv. Det gjenstår å undersøke om epigenetiske endringer ved alvorlige psykiske lidelser sammenfaller med de som ses etter psykososialt stress tidlig i livet.
Teksten sto på trykk første gang i Tidsskrift for Norsk psykologforening, Vol 55, nummer 10, 2017, side 940-950
Referanser
Abdolmaleky, H. M., Cheng, K. H., Russo, A., Smith, C. L., Faraone, S. V., Wilcox, M., . . . Tsuang, M. T. (2005). Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet, 134B(1), 60–66. doi:10.1002/ajmg.b.30140
Abdolmaleky, H. M., Nohesara, S., Ghadirivasfi, M., Lambert, A. W., Ahmadkhaniha, H., Ozturk, S., . . . Thiagalingam, S. (2014). DNA hypermethylation of serotonin transporter gene promoter in drug naive patients with schizophrenia. Schizophr Res, 152(2–3), 373–380. doi:10.1016/j.schres.2013.12.007
Abdolmaleky, H. M., Yaqubi, S., Papageorgis, P., Lambert, A. W., Ozturk, S., Sivaraman, V., & Thiagalingam, S. (2011). Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr Res, 129(2–3), 183–190. doi:10.1016/j.schres.2011.04.007
Antequera, F., & Bird, A. (1993). Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A, 90(24), 11995–11999.
Bakulski, K. M., Halladay, A., Hu, V. W., Mill, J., & Fallin, M. D. (2016). Epigenetic Research in Neuropsychiatric Disorders: the «Tissue Issue». Curr Behav Neurosci Rep, 3(3), 264–274. doi:10.1007/s40473–016–0083–4
Bock, J., Rether, K., Groger, N., Xie, L., & Braun, K. (2014). Perinatal programming of emotional brain circuits: an integrative view from systems to molecules. Front Neurosci, 8, 11. doi:10.3389/fnins.2014.00011
Cariaga-Martinez, A., & Alelu-Paz, R. (2016). False data, positive results in neurobiology: moving beyond the epigenetics of blood and saliva samples in mental disorders. J Negat Results Biomed, 15(1), 21. doi:10.1186/s12952–016–0064-x
Caspi, A., Sugden, K., Moffitt, T. E., Taylor, A., Craig, I. W., Harrington, H., . . . Poulton, R. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science, 301(5631), 386–389. doi:10.1126/science.1083968
Cheah, S. Y., McLeay, R., Wockner, L. F., Lawford, B. R., Young, R. M., Morris, C. P., & Voisey, J. (2016). Expression and methylation of BDNF in the human brain in schizophrenia. World J Biol Psychiatry, 1–9. doi:10.1080/15622975.2016.1245443
Chiba, S., Numakawa, T., Ninomiya, M., Richards, M. C., Wakabayashi, C., & Kunugi, H. (2012). Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry, 39(1), 112–119. doi:10.1016/j.pnpbp.2012.05.018
Davies, M. N., Volta, M., Pidsley, R., Lunnon, K., Dixit, A., Lovestone, S., . . . Mill, J. (2012). Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol, 13(6), R43. doi:10.1186/gb-2012–13–6-r43
Deak, T., Quinn, M., Cidlowski, J. A., Victoria, N. C., Murphy, A. Z., & Sheridan, J. F. (2015). Neuroimmune mechanisms of stress: sex differences, developmental plasticity, and implications for pharmacotherapy of stress-related disease. Stress, 18(4), 367–380. doi:10.3109/10253890.2015.1053451
Deans, C., & Maggert, K. A. (2015). What do you mean, «epigenetic»? Genetics, 199(4), 887–896. doi:10.1534/genetics.114.173492
Encode Project Consortium. (2007). Identification and analysis of functional elements in 1 % of the human genome by the ENCODE pilot project. Nature, 447(7146), 799–816. doi:10.1038/nature05874
Goldberg, A. D., Allis, C. D., & Bernstein, E. (2007). Epigenetics: a landscape takes shape. Cell, 128(4), 635–638. doi:10.1016/j.cell.2007.02.006
Griffiths, B. B., & Hunter, R. G. (2014). Neuroepigenetics of stress. Neuroscience, 275, 420–435. doi:10.1016/j.neuroscience.2014.06.041
Haghighi, F., Xin, Y., Chanrion, B., O’Donnell, A. H., Ge, Y., Dwork, A. J., . . . Mann, J. J. (2014). Increased DNA methylation in the suicide brain. Dialogues Clin Neurosci, 16(3), 430–438.
Hannon, E., Dempster, E., Viana, J., Burrage, J., Smith, A. R., Macdonald, R., . . . Mill, J. (2016). An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol, 17(1), 176. doi:10.1186/s13059–016–1041-x
Heijmans, B. T., & Mill, J. (2012). Commentary: The seven plagues of epigenetic epidemiology. Int J Epidemiol, 41(1), 74–78. doi:10.1093/ije/dyr225
Herman, J. P., McKlveen, J. M., Ghosal, S., Kopp, B., Wulsin, A., Makinson, R., . . . Myers, B. (2016). Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr Physiol, 6(2), 603–621. doi:10.1002/cphy.c150015
Holly, E. N., & Miczek, K. A. (2016). Ventral tegmental area dopamine revisited: effects of acute and repeated stress. Psychopharmacology (Berl), 233(2), 163–186. doi:10.1007/s00213–015–4151–3
Jaffe, A. E. (2016). Postmortem human brain genomics in neuropsychiatric disorders--how far can we go? Curr Opin Neurobiol, 36, 107–111. doi:10.1016/j.conb.2015.11.002
Jaffe, A. E., Gao, Y., Deep-Soboslay, A., Tao, R., Hyde, T. M., Weinberger, D. R., & Kleinman, J. E. (2016). Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci, 19(1), 40–47. doi:10.1038/nn.4181
Jaffe, A. E., & Kleinman, J. E. (2016). Genetic and epigenetic analysis of schizophrenia in blood-a no-brainer? Genome Med, 8(1), 96. doi:10.1186/s13073–016–0354–4
Jawahar, M. C., Murgatroyd, C., Harrison, E. L., & Baune, B. T. (2015). Epigenetic alterations following early postnatal stress: a review on novel aetiological mechanisms of common psychiatric disorders. Clin Epigenetics, 7, 122. doi:10.1186/s13148–015–0156–3
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet, 13(7), 484–492. doi:10.1038/nrg3230
Keller, S., Errico, F., Zarrilli, F., Florio, E., Punzo, D., Mansueto, S., . . . Chiariotti, L. (2014). DNA methylation state of BDNF gene is not altered in prefrontal cortex and striatum of schizophrenia subjects. Psychiatry Res, 220(3), 1147–1150. doi:10.1016/j.psychres.2014.08.022
Koolhaas, J. M., Bartolomucci, A., Buwalda, B., de Boer, S. F., Flugge, G., Korte, S. M., . . . Fuchs, E. (2011). Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev, 35(5), 1291–1301. doi:10.1016/j.neubiorev.2011.02.003
Kozlenkov, A., Wang, M., Roussos, P., Rudchenko, S., Barbu, M., Bibikova, M., . . . Dracheva, S. (2016). Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res, 44(6), 2593–2612. doi:10.1093/nar/gkv1304
Kundakovic, M., & Champagne, F. A. (2015). Early-life experience, epigenetics, and the developing brain. Neuropsychopharmacology, 40(1), 141–153. doi:10.1038/npp.2014.140
Kundakovic, M., & Jaric, I. (2017). The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders. Genes (Basel), 8(3). doi:10.3390/genes8030104
Labonte, B., Suderman, M., Maussion, G., Navaro, L., Yerko, V., Mahar, I., . . . Turecki, G. (2012). Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry, 69(7), 722–731. doi:10.1001/archgenpsychiatry.2011.2287
Le Francois, B., Soo, J., Millar, A. M., Daigle, M., Le Guisquet, A. M., Leman, S., . . . Albert, P. R. (2015). Chronic mild stress and antidepressant treatment alter 5-HT1A receptor expression by modifying DNA methylation of a conserved Sp4 site. Neurobiol Dis, 82, 332–341. doi:10.1016/j.nbd.2015.07.002
Lomazzo, E., Konig, F., Abassi, L., Jelinek, R., & Lutz, B. (2017). Chronic stress leads to epigenetic dysregulation in the neuropeptide-Y and cannabinoid CB1 receptor genes in the mouse cingulate cortex. Neuropharmacology, 113(Pt A), 301–313. doi:10.1016/j.neuropharm.2016.10.008
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., & Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature, 389(6648), 251–260. doi:10.1038/38444
Matrisciano, F., Tueting, P., Dalal, I., Kadriu, B., Grayson, D. R., Davis, J. M., . . . Guidotti, A. (2013). Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology, 68, 184–194. doi:10.1016/j.neuropharm.2012.04.013
McEwen, B. S., Bowles, N. P., Gray, J. D., Hill, M. N., Hunter, R. G., Karatsoreos, I. N., & Nasca, C. (2015). Mechanisms of stress in the brain. Nat Neurosci, 18(10), 1353–1363. doi:10.1038/nn.4086
McGowan, P. O., Sasaki, A., D’Alessio, A. C., Dymov, S., Labonte, B., Szyf, M., . . . Meaney, M. J. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci, 12(3), 342–348. doi:10.1038/nn.2270
McGowan, P. O., & Szyf, M. (2010). The epigenetics of social adversity in early life: implications for mental health outcomes. Neurobiol Dis, 39(1), 66–72. doi:10.1016/j.nbd.2009.12.026
Meaney, M. J., & Szyf, M. (2005). Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci, 7(2), 103–123.
Meissner, A. (2010). Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol, 28(10), 1079–1088. doi:10.1038/nbt.1684
Mill, J., Tang, T., Kaminsky, Z., Khare, T., Yazdanpanah, S., Bouchard, L., . . . Petronis, A. (2008). Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet, 82(3), 696–711. doi:10.1016/j.ajhg.2008.01.008
Mitchell, A., Roussos, P., Peter, C., Tsankova, N., & Akbarian, S. (2014). The future of neuroepigenetics in the human brain. Prog Mol Biol Transl Sci, 128, 199–228. doi:10.1016/B978–0–12–800977–2.00008–5
Monoranu, C. M., Grunblatt, E., Bartl, J., Meyer, A., Apfelbacher, M., Keller, D., . . . Riederer, P. (2011). Methyl- and acetyltransferases are stable epigenetic markers postmortem. Cell Tissue Bank, 12(4), 289–297. doi:10.1007/s10561–010–9199-z
Mueller, B. R., & Bale, T. L. (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci, 28(36), 9055–9065. doi:10.1523/JNEUROSCI.1424–08.2008
Murphy, T. M., Crawford, B., Dempster, E. L., Hannon, E., Burrage, J., Turecki, G., . . . Mill, J. (2017). Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl Psychiatry, 7(1), e989. doi:10.1038/tp.2016.249
Mychasiuk, R., Ilnytskyy, S., Kovalchuk, O., Kolb, B., & Gibb, R. (2011). Intensity matters: brain, behaviour and the epigenome of prenatally stressed rats. Neuroscience, 180, 105–110. doi:10.1016/j.neuroscience.2011.02.026
Nestler, E. J., Pena, C. J., Kundakovic, M., Mitchell, A., & Akbarian, S. (2015). Epigenetic Basis of Mental Illness. Neuroscientist. doi:10.1177/1073858415608147
Nordanger, D. Ø., & Braarud, H. C. (2017). Utviklingstraumer. Regulering som nøkkelbegrep i en ny traumepsykologi. Bergen: Fagbokforlaget.
Numata, S., Ye, T., Herman, M., & Lipska, B. K. (2014). DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia. Front Genet, 5, 280. doi:10.3389/fgene.2014.00280
Palacios-Garcia, I., Lara-Vasquez, A., Montiel, J. F., Diaz-Veliz, G. F., Sepulveda, H., Utreras, E., . . . Aboitiz, F. (2015). Prenatal stress down-regulates Reelin expression by methylation of its promoter and induces adult behavioral impairments in rats. PLoS One, 10(2), e0117680. doi:10.1371/journal.pone.0117680
Patel, P. D., Katz, M., Karssen, A. M., & Lyons, D. M. (2008). Stress-induced changes in corticosteroid receptor expression in primate hippocampus and prefrontal cortex. Psychoneuroendocrinology, 33(3), 360–367. doi:10.1016/j.psyneuen.2007.12.003
Pidsley, R., Viana, J., Hannon, E., Spiers, H., Troakes, C., Al-Saraj, S., . . . Mill, J. (2014). Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol, 15(10), 483. doi:10.1186/s13059–014–0483–2
Provencal, N., Suderman, M. J., Guillemin, C., Massart, R., Ruggiero, A., Wang, D., . . . Szyf, M. (2012). The signature of maternal rearing in the methylome in rhesus macaque prefrontal cortex and T cells. J Neurosci, 32(44), 15626–15642. doi:10.1523/JNEUROSCI.1470–12.2012
Pruessner, M., Cullen, A. E., Aas, M., & Walker, E. F. (2017). The neural diathesis-stress model of schizophrenia revisited: An update on recent findings considering illness stage and neurobiological and methodological complexities. Neurosci Biobehav Rev, 73, 191–218. doi:10.1016/j.neubiorev.2016.12.013
Read, J., Fosse, R., Moskowitz, A., & Perry, B. D. (2014). The traumagenic neurodevelopmental model of psychosis revisited. Neuropsychiatry, 4(1), 65–79.
Roadmap Epigenomics, C., Kundaje, A., Meuleman, W., Ernst, J., Bilenky, M., Yen, A., . . . Kellis, M. (2015). Integrative analysis of 111 reference human epigenomes. Nature, 518(7539), 317–330. doi:10.1038/nature14248
Sabunciyan, S., Aryee, M. J., Irizarry, R. A., Rongione, M., Webster, M. J., Kaufman, W. E., . . . Gen, R. E. D. C. (2012). Genome-wide DNA methylation scan in major depressive disorder. PLoS One, 7(4), e34451. doi:10.1371/journal.pone.0034451
Saxonov, S., Berg, P., & Brutlag, D. L. (2006). A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A, 103(5), 1412–1417. doi:10.1073/pnas.0510310103
Selye, H. (1974). Stress without distress. Philadelphia: J.B. Lippincott Company.
Tang, B., Dean, B., & Thomas, E. A. (2011). Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry, 1, e64. doi:10.1038/tp.2011.61
Teicher, M. H., & Samson, J. A. (2016). Annual Research Review: Enduring neurobiological effects of childhood abuse and neglect. J Child Psychol Psychiatry, 57(3), 241–266. doi:10.1111/jcpp.12507
Tochigi, M., Iwamoto, K., Bundo, M., Komori, A., Sasaki, T., Kato, N., & Kato, T. (2008). Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry, 63(5), 530–533. doi:10.1016/j.biopsych.2007.07.003
Tung, J., & Gilad, Y. (2013). Social environmental effects on gene regulation. Cell Mol Life Sci, 70(22), 4323–4339. doi:10.1007/s00018–013–1357–6
Uchida, T., Furukawa, T., Iwata, S., Yanagawa, Y., & Fukuda, A. (2014). Selective loss of parvalbumin-positive GABAergic interneurons in the cerebral cortex of maternally stressed Gad1-heterozygous mouse offspring. Transl Psychiatry, 4, e371. doi:10.1038/tp.2014.13
Veeraiah, P., Noronha, J. M., Maitra, S., Bagga, P., Khandelwal, N., Chakravarty, S., . . . Patel, A. B. (2014). Dysfunctional glutamatergic and gamma-aminobutyric acidergic activities in prefrontal cortex of mice in social defeat model of depression. Biol Psychiatry, 76(3), 231–238. doi:10.1016/j.biopsych.2013.09.024
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., . . . Zhu, X. (2001). The sequence of the human genome. Science, 291(5507), 1304–1351. doi:10.1126/science.1058040
Viana, J., Hannon, E., Dempster, E., Pidsley, R., Macdonald, R., Knox, O., . . . Mill, J. (2016). Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum Mol Genet. doi:10.1093/hmg/ddw373
Waddington, C. H. (1942). The epigenotype. Endeavour, 1, 18–20.
Walker, E. F., & Diforio, D. (1997). Schizophrenia: a neural diathesis-stress model. Psychol Rev, 104(4), 667–685.
Walton, E., Hass, J., Liu, J., Roffman, J. L., Bernardoni, F., Roessner, V., . . . Ehrlich, S. (2016). Correspondence of DNA Methylation Between Blood and Brain Tissue and Its Application to Schizophrenia Research. Schizophr Bull, 42(2), 406–414. doi:10.1093/schbul/sbv074
Weaver, I. C. (2009). Shaping adult phenotypes through early life environments. Birth Defects Res C Embryo Today, 87(4), 314–326. doi:10.1002/bdrc.20164
Weaver, I. C. (2014). Integrating early life experience, gene expression, brain development, and emergent phenotypes: unraveling the thread of nature via nurture. Adv Genet, 86, 277–307. doi:10.1016/B978–0–12–800222–3.00011–5
Weaver, I. C., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., . . . Meaney, M. J. (2004). Epigenetic programming by maternal behavior. Nat Neurosci, 7(8), 847–854. doi:10.1038/nn1276
Weickert, C. S., Hyde, T. M., Lipska, B. K., Herman, M. M., Weinberger, D. R., & Kleinman, J. E. (2003). Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry, 8(6), 592–610. doi:10.1038/sj.mp.4001308
Wockner, L. F., Noble, E. P., Lawford, B. R., Young, R. M., Morris, C. P., Whitehall, V. L., & Voisey, J. (2014). Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry, 4, e339. doi:10.1038/tp.2013.111
Xiao, Y., Camarillo, C., Ping, Y., Arana, T. B., Zhao, H., Thompson, P. M., . . . Xu, C. (2014). The DNA methylome and transcriptome of different brain regions in schizophrenia and bipolar disorder. PLoS One, 9(4), e95875. doi:10.1371/journal.pone.0095875
Zhang, T. Y., Labonte, B., Wen, X. L., Turecki, G., & Meaney, M. J. (2013). Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology, 38(1), 111–123. doi:10.1038/npp.2012.149
")
